Importing Spatial Data
Importing Spatial Datasets
VoltRon is an end-to-end spatial omics analysis package that supports a large selection of spatial data resolutions. Currently, there exists a considerable amount of spatial omics technologies that generate datasets whose omics profiles are spatially resolved.
VoltRon objects are compatible with readouts of almost all of these technologies where we provide a selection of built-in functions to help users constructing VoltRon objects with ease. In this tutorial, we will review these spatial omics instruments and the functions available within the VoltRon package to import their readouts.
Visium (10x Genomics)
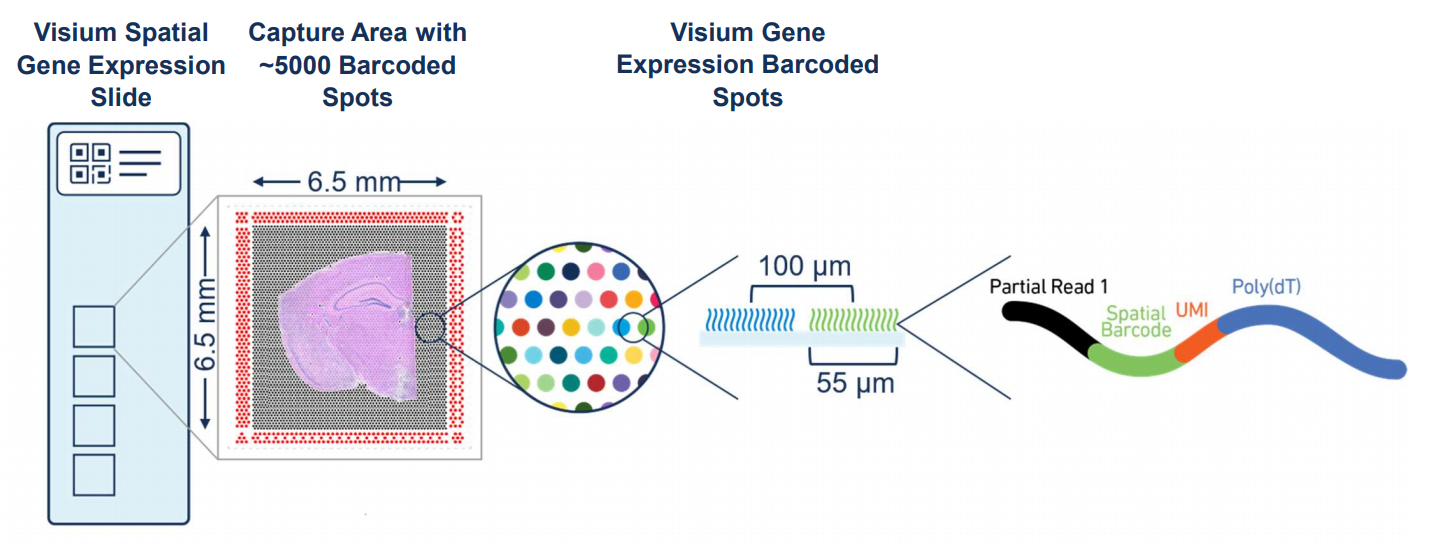
Image Credit: The Visium Spatial Gene Expression Slide (https://www.10xgenomics.com/)
10x Genomics Visium Spatial Gene Expression Platform incorporates in situ arrays (i.e. spots) to capture spatial localization of omics profiles where these spots are of 55 \(\mu\)m in diameter and constitute a grid that covers a significant portion of a tissue section placed on the slide of the instrument.
We will use the readouts of Visium CytAssist platform that was derived from a single tissue section of a breast cancer sample. More information on the Visium CytAssist data and the study can also be found on the bioRxiv preprint. You can download the data from the 10x Genomics website (specifically, import the Visium Spatial data).
Before we start, Visium readouts require rhdf5 package to be installed.
if(!requireNamespace("rhdf5"))
BiocManager::install("rhdf5")We use the importVisium function to import the Visium readouts and create a VoltRon object. Here, we point to the folder of all the files with dir.path argument and also determine the name of this sample (sample_name).
library(VoltRon)
Vis_R1 <- importVisium(dir.path = "Visium/", sample_name = "VisiumR1")VoltRon Object
VisiumR1:
Layers: Section1
Assays: Visium(Main) While importing the readouts, we can also determine the name of the assay as well as the name of the image. The SampleMetadata function summarizes the entire collection of assays, layers (sections) and samples (tissue blocks) within the R object.
Vis_R1 <- importVisium(dir.path = "Visium/", sample_name = "VisiumR1",
assay_name = "Visium_assay", image_name = "H&E_stain")
SampleMetadata(Vis_R1) Assay Layer Sample
Assay1 Visium_assay Section1 VisiumR1The current VoltRon object has only one assay associated with a single layer and a tissue block, and the image of this assay is currently the “H&E_stain”.
vrImageNames(Vis_R1)[1] "H&E_stain"Although by default the importVisium function selects the low resolution image, you can select the higher resolution one using resolution_level=“hires”
Vis_R1 <- importVisium(dir.path = "Visium/", sample_name = "VisiumR1", resolution_level="hires")VisiumHD (10x Genomics)
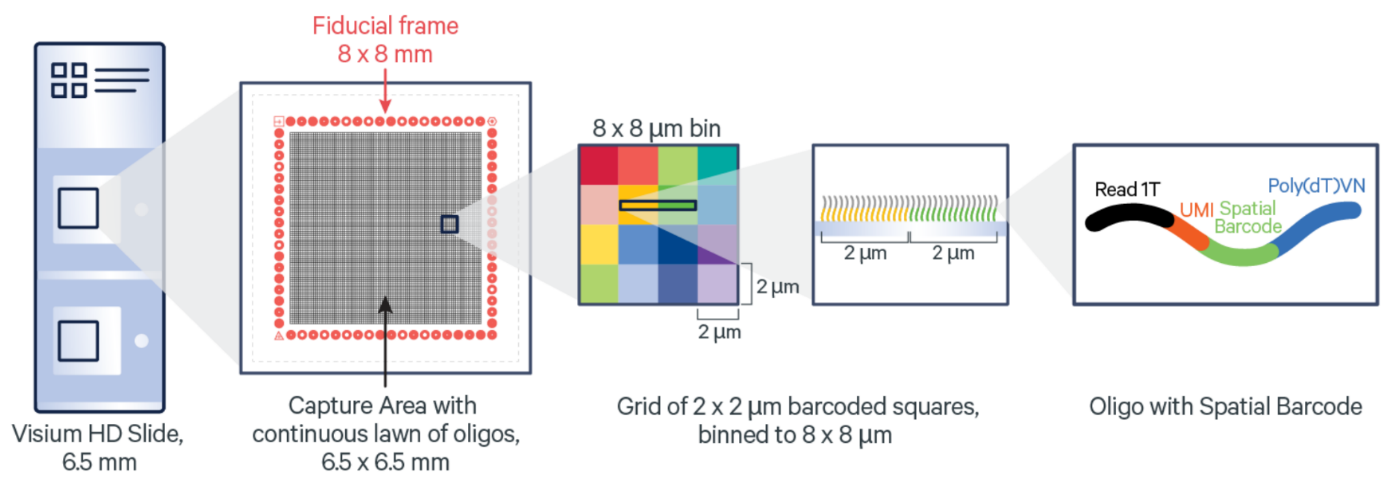
https://www.10xgenomics.com/products/visium-hd-spatial-gene-expression
10x Genomics VisiumHD Spatial Gene Expression Platform contains two 6.5 x 6.5 mm Capture Areas with a continuous lawn of oligonucleotides arrayed in millions of 2 x 2 µm barcoded squares without gaps, achieving single cell–scale spatial resolution. The data is output at 2 µm, as well as multiple bin sizes. The 8 x 8 µm bin is the recommended starting point for visualization and analysis.
We will use the readouts of Visium CytAssist platform that was derived from a single tissue section of a breast cancer sample. More information on the Visium CytAssist data and the study can also be found on the bioRxiv preprint. You can download the data from the 10x Genomics website (specifically, import the Visium Spatial data).
Before we start, VisiumHD readouts require a number of Bioconductor and R packages to be installed.
if(!requireNamespace("arrow"))
install.packages("arrow")
if(!requireNamespace("rhdf5"))
BiocManager::install("rhdf5")We use the importVisiumHD function to import the Visium readouts and create a VoltRon object. Here, we point to the folder of all the files with dir.path argument and also determine the name of this sample (sample_name).
library(VoltRon)
hddata <- importVisiumHD(dir.path = "VisiumHD/outs/",
sample_name = "VisiumHD")The VisiumHD readouts provide multiple bin sizes which are aggregated versions of the original 2\(\mu\)m \(x\) 2\(\mu\)m capture spots. The default bin sizes are (i) 2\(\mu\)m \(x\) 2\(\mu\)m, (ii) 8\(\mu\)m \(x\) 8\(\mu\)m and (iii) 16\(\mu\)m \(x\) 16\(\mu\)m. Although by default the importVisium function selects the low resolution image, you can select the higher resolution one using resolution_level=“hires”.
hddata <- importVisiumHD(dir.path = "VisiumHD/outs/",
bin.size = "16",
resolution_level = "hires",
sample_name = "VisiumHD")Xenium (10x Genomics)
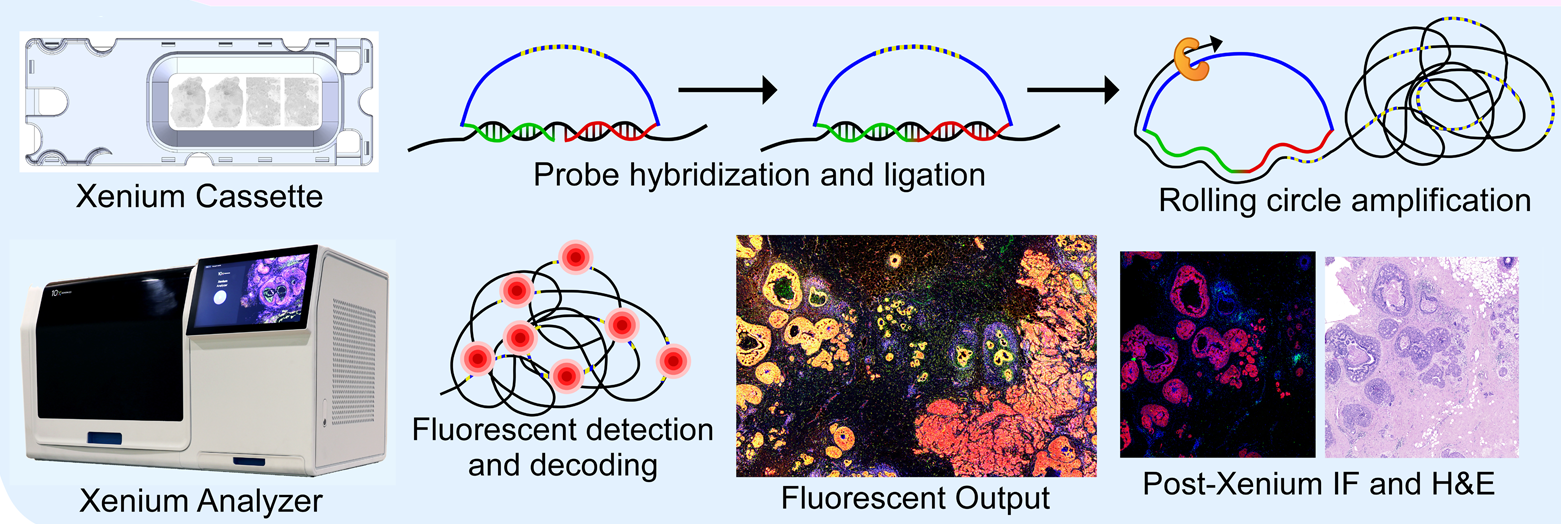
Image Credit: https://www.biorxiv.org/content/10.1101/2022.10.06.510405v2
The 10x Genomics Xenium In Situ provides spatial localization of both (i) transcripts from a few hundred genes as well as (ii) the single cells with transcriptomics profiles.
We will use the readouts of a single Xenium platform replicate that was derived from a single tissue section of a breast cancer sample. More information on the Xenium data and the study can also be found on the bioRxiv preprint. You can download the data from the 10x Genomics website (specifically, import the In Situ Replicate 1 data).
Before we start, Xenium readouts require a number of Bioconductor and R packages to be installed. RBioFormats requires rJava and JDK installed, see dependencies section for more information.
if(!requireNamespace("RBioFormats"))
BiocManager::install("RBioFormats")
if(!requireNamespace("arrow"))
install.packages("arrow")
if(!requireNamespace("rhdf5"))
BiocManager::install("rhdf5")We use the importXenium function to import the Xenium readouts and create a VoltRon object. Here, we point to the folder of all the files with dir.path argument and also determine the name of this sample (sample_name).
library(VoltRon)
Xen_R1 <- importXenium("Xenium_R1/outs", sample_name = "XeniumR1")VoltRon Object
XeniumR1:
Layers: Section1
Assays: Xenium(Main) You can use the import_molecules argument to import positions and features of the transcripts along with the single cell profiles.
Xen_R1 <- importXenium("Xenium_R1/outs", sample_name = "XeniumR1", import_molecules = TRUE)
Xen_R1VoltRon Object
XeniumR1:
Layers: Section1
Assays: Xenium(Main) Xenium_mol The SampleMetadata function summarizes the entire collection of assays, layers (sections) and samples (tissue blocks) within the R object. In this case, the function will generate two assays in a single layer where one is a “cell” assay and the other is a “molecule assay”.
SampleMetadata(Xen_R1) Assay Layer Sample
Assay1 Xenium Section1 XeniumR1
Assay2 Xenium_mol Section1 XeniumR1The Xenium in situ platform provides multiple resolution of the same Xenium slide which can be parsed from the OME-TIFF image file of DAPI stained tissue section (e.g. morphology_mip.ome.tif). The resolution_level argument determines the resolution of the DAPI image generated from the OME-TIFF file. More information on resolution levels can be found here.
Xen_R1 <- importXenium("Xenium_R1/outs", sample_name = "XeniumR1", import_molecules = TRUE,
resolution_level = 4, overwrite_resolution = TRUE)
vrImages(Xen_R1, assay = "Xenium")# A tibble: 1 × 7
format width height colorspace matte filesize density
<chr> <int> <int> <chr> <lgl> <int> <chr>
1 PNG 4427 3222 Gray FALSE 0 72x72 Users can also decide to ignore OME-TIFF file and images, hence only cells and molecules would be imported.
Xen_R1 <- importXenium("Xenium_R1/outs", sample_name = "XeniumR1", import_molecules = TRUE,
use_image = FALSE)GeoMx (NanoString)

Image Credit: https://www.biochain.com/nanostring-geomx-digital-spatial-profiling/
The NanoString’s GeoMx Digital Spatial Profiler is a high-plex spatial profiling technology which produces segmentation-based protein and RNA assays. The instrument allows users to select regions of interest (ROIs) from fluorescent microscopy images that capture the morphological context of the tissue. These are ROIs are then used to generate transcriptomics or proteomics profiles.
We will import the ROI profiles generated from the GeoMx scan area where COVID-19 lung tissues were fitted into. See GSE190732 for more information on this study.
Here is the usage of importGeoMx function and necessary files for this example:
| Argument | Description | Link |
|---|---|---|
| dcc.path | The path to DCC files directory | DDC files |
| pkc.file | GeoMx™ DSP configuration file | Human RNA Whole Transcriptomics Atlas for NGS |
| summarySegment | Segment summary table (.xls or .csv) | ROI Metadata file |
| image | The Morphology Image of the scan area | Image file |
| ome.tiff | The OME-TIFF Image of the scan area | OME-TIFF file |
| The OME-TIFF Image XML file | OME-TIFF (XML) file |
library(VoltRon)
GeoMxR1 <- importGeoMx(dcc.path = "DCC-20230427/",
pkc.file = "Hs_R_NGS_WTA_v1.0.pkc",
summarySegment = "segmentSummary.csv",
image = "Lu1A1B5umtrueexp.tif",
ome.tiff = "Lu1A1B5umtrueexp.ome.tiff",
sample_name = "GeoMxR1")The OME-TIFF file here provides the ROI coordinates within the embedded XML metadata. We can also incorporate the RBioFormats package to extract the XML metadata from the OME-TIFF file. RBioFormats requires rJava and JDK installed, see dependencies section for more information.
if(!requireNamespace("RBioFormats"))
BiocManager::install("RBioFormats")
library(RBioFormats)
# alternatively you can use RBioFormats to create an xml file
ome.tiff.xml <- RBioFormats::read.omexml("data/GeoMx/Lu1A1B5umtrueexp.ome.tiff")
write(ome.tiff.xml, file = "data/GeoMx/Lu1A1B5umtrueexp.ome.tiff.xml")The ome.tiff argument also accepts the path to this XML file.
GeoMxR1 <- importGeoMx(dcc.path = "DCC-20230427/",
pkc.file = "Hs_R_NGS_WTA_v1.0.pkc",
summarySegment = "segmentSummary.csv",
image = "Lu1A1B5umtrueexp.tif",
ome.tiff = "Lu1A1B5umtrueexp.ome.tiff.xml",
sample_name = "GeoMxR1")CosMx (NanoString)
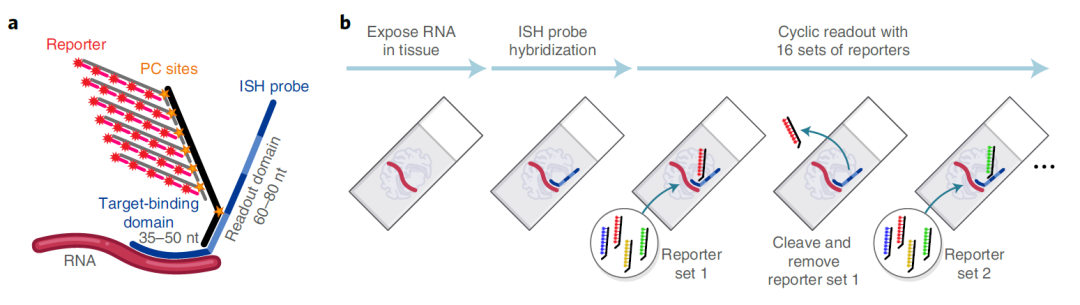
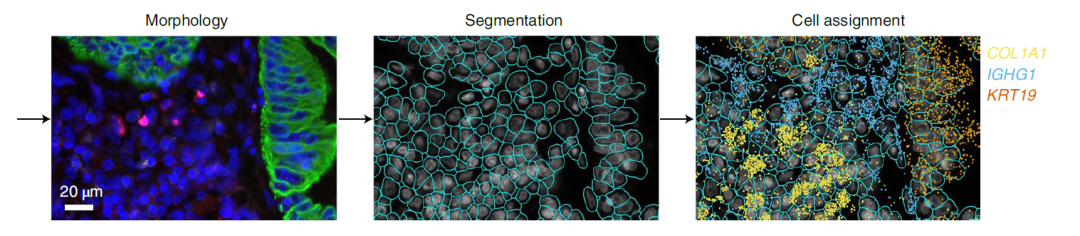
Image Credit: https://www.biorxiv.org/content/10.1101/2021.11.03.467020v1.full
The NanoString’s CosMx Spatial Molecular Imaging platform is a high-plex spatial multiomics technology that captures the spatial localization of both (i) transcripts from thousands of genes as well as (ii) the single cells with transcriptomics and proteomics profiles.
We will use the readouts from two slides of a single CosMx experiment. You can download the data from the NanoString website.
We use the importCosMx function to import the CosMx readouts and create a VoltRon object. Here, we point to the folder of the TileDB array that stores feature matrix as well as the transcript metadata.
CosMxR1 <- importCosMx(tiledbURI = "MuBrainDataRelease/")VoltRon Object
Slide1:
Layers: Section1
Slide2:
Layers: Section1
Assays: CosMx(Main) You can use the import_molecules argument to import positions and features of the transcripts along with the single cell profiles.
CosMxR1 <- importCosMx(tiledbURI = "MuBrainDataRelease/", import_molecules = TRUE)STOmics (MGI)
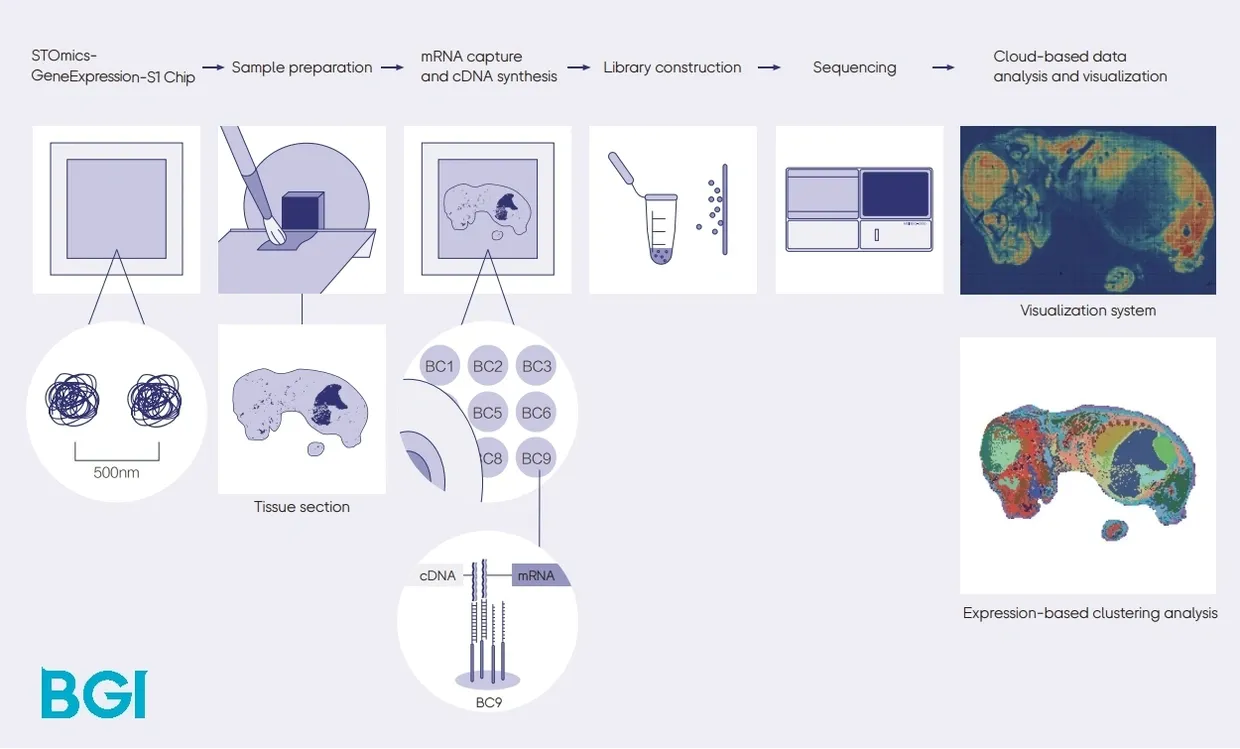
Image Credit: https://bgi-australia.com.au/stomics
Before importing the STOmics data to VoltRon, we first convert STOmics readouts to an h5ad file using the stereopy Python module. For more information, visit https://stereopy.readthedocs.io/. See here for instructions on how to install stereopy.
import stereo as st
import warnings
warnings.filterwarnings('ignore')
# read the GEF file
data_path = './SS200000135TL_D1.tissue.gef'
data = st.io.read_gef(file_path=data_path, bin_size=50)
data.tl.raw_checkpoint()
# remember to set flavor as scanpy
adata = st.io.stereo_to_anndata(data, flavor='scanpy', output='sample.h5ad')We use the importSTOmics function to import the STOmics readouts and create a VoltRon object. Here, we point to the folder an h5ad file generated using the stereo.io.stereo_to_anndata method previously.
vrdata <- importSTOmics(h5ad.path = "sample.h5ad")VoltRon Object
Sample1:
Layers: Section1
Assays: STOmics(Main) GenePS (Spatial Genomics)
Image Credit: https://spatialgenomics.com/applications/
We use the importGenePS function to import the GenePS (Spatial Genomics) readouts and create a VoltRon object. You can use the import_molecules argument to import positions and features of the transcripts along with the single cell profiles. The resolution_level argument determines the resolution of the DAPI image generated from the TIFF file.
vrdata <- importGenePS(dir.path = "out/", import_molecules = TRUE, resolution_level = 7)VoltRon Object
Sample1:
Layers: Section1
Assays: GenePS(Main) GenePS_molPhenoCycler (Akoya)

Image Credit: https://tep.cancer.illinois.edu/phenocycler-fusion-system/
We use the importPhenoCycler function to import the PhenoCycler (Akoya Biosciences) readouts and create a VoltRon object. The function supports multiple readouts types depending on how the readouts were generated, this is controlled by the type arguement. For more information on all arguements of the function, see help(importPhenoCycler).
We used the Human FFPE tonsil tissue example with 83000 cells which could be found here. You have to download the contents and dir.path arguement should be set to the location of the Example-dataset-for-MAV folder.
vr_pheno <- importPhenoCycler(dir.path = "Example-dataset-for-MAV/", type = "processor",
sample_name = "Tonsil18AB")VoltRon Object
Tonsil18AB:
Layers: Section1
Assays: PhenoCycler(Main) OpenST
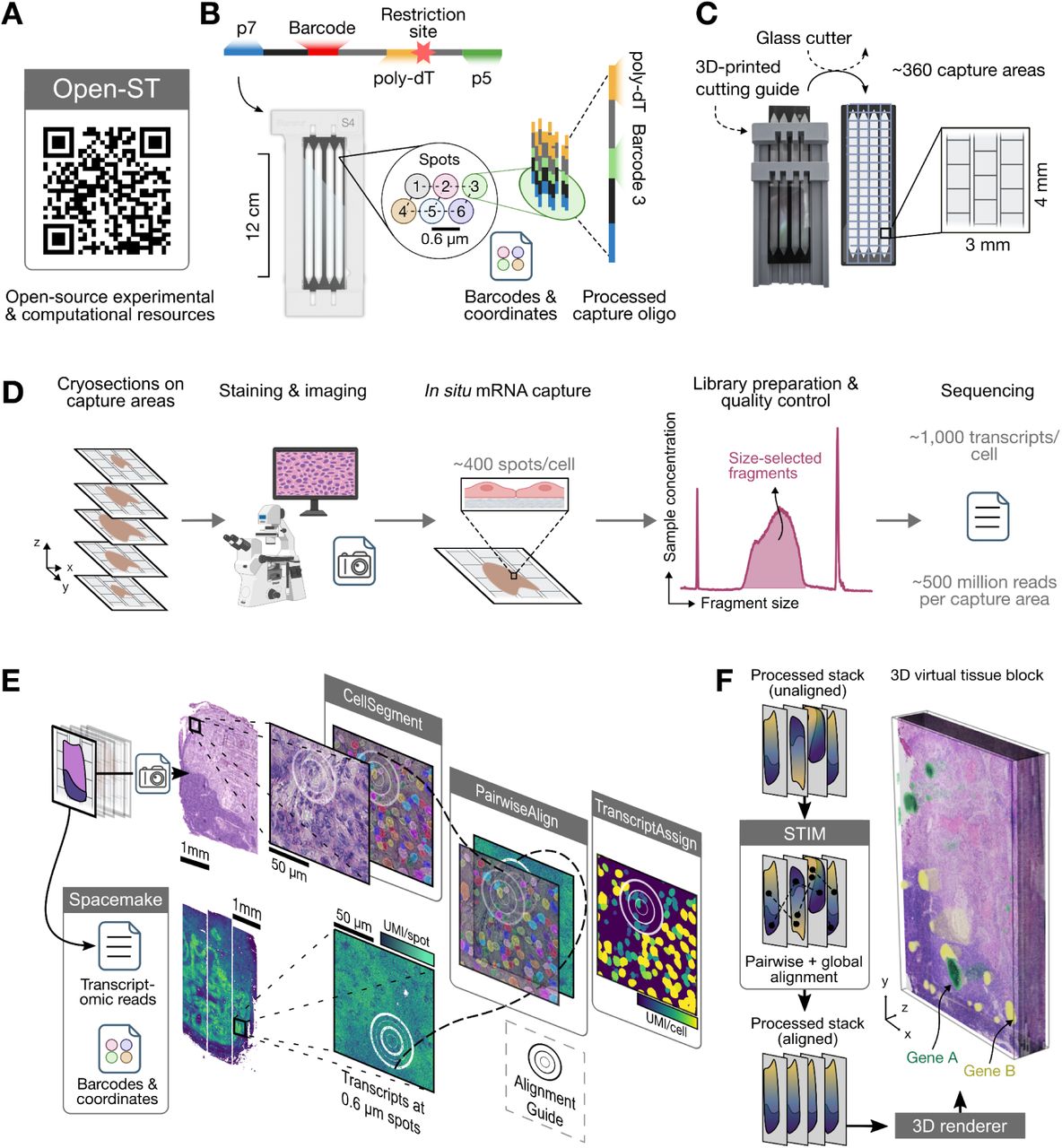
Image Credit: https://www.cell.com/cell/fulltext/S0092-8674(24)00636-6
We use the importOpenST function to import the OpenST https://rajewsky-lab.github.io/openst/latest/ readouts and create a VoltRon object. The function will parse each section from the output h5ad file, define it as an independent assay in a single layer where these layers are ordered in a single tissue block.
We use the metastatic lymph node example that is deposited to NCBI/GEO (GSE251926). Please use the GSE251926_metastatic_lymph_node_3d.h5ad.gz file and unzip it to use the importOpenST function below.
vr_openst <- importOpenST(h5ad.path = "GSE251926_metastatic_lymph_node_3d.h5ad",
sample_name = "MLN_3D")VoltRon Object
MLN_3D:
Layers: Section1 Section2 Section3 Section4 Section5 Section6 Section7 Section8 Section9 Section10 Section11 Section12 Section13 Section14 Section15 Section16 Section17 Section18 Section19
Assays: OpenST(Main) DBIT-Seq
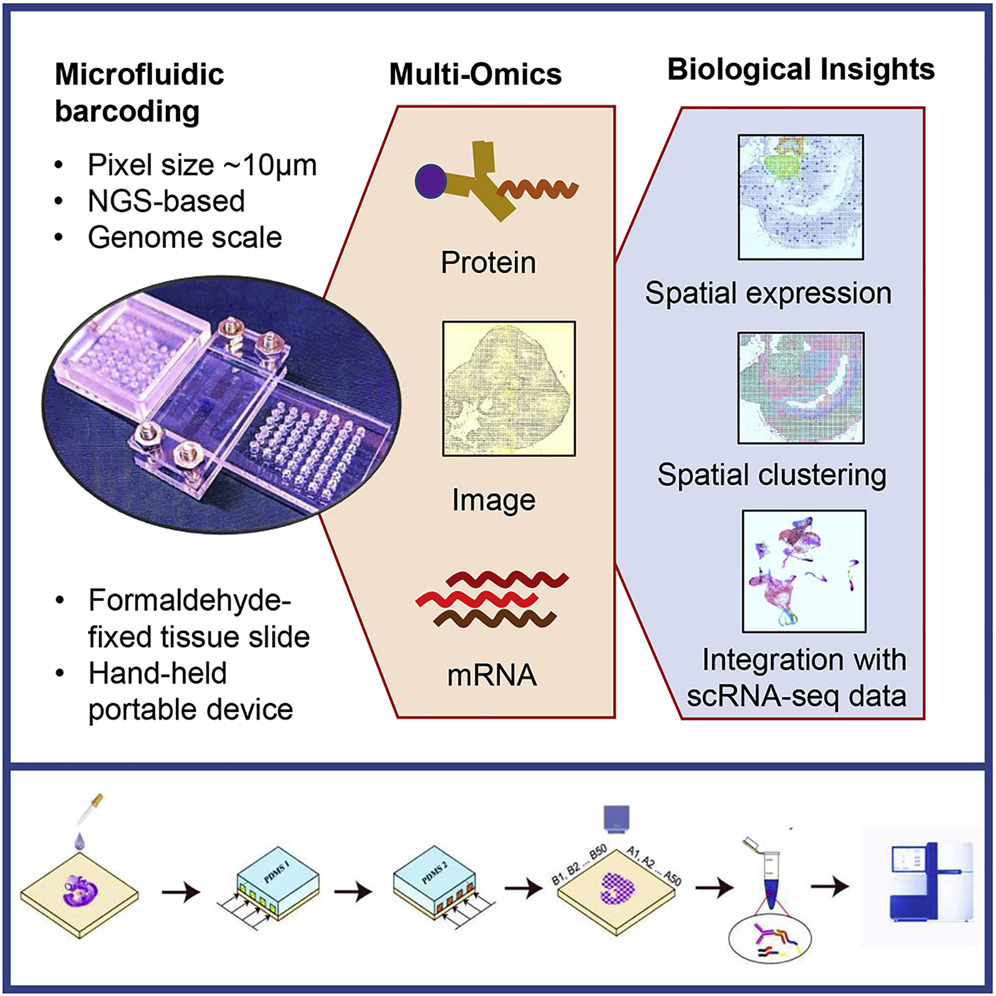
Image Credit: https://www.cell.com/cell/fulltext/S0092-8674(20)31390-8
We use the importDBITSeq function to import the DBIT-Seq https://www.cell.com/cell/fulltext/S0092-8674(20)31390-8 readouts and create a VoltRon object. The default path to the rna count matrix is accompanied by the path to the protein count matrix, which is optional. The size parameter here determines the size of each square pixel on the DBIT-Seq slide (default is 10\(\mu\)m).
We use the example with developing eye field in a E10 mouse embryo using 10-μm microfluidic (sample id: 0719cL for RNA, and 0719aL for Protein) channels that is deposited to NCBI/GEO (GSE137986).
vr_dbit <- importDBITSeq(path.rna = "GSM4189615_0719cL.tsv",
path.prot = "GSM4202309_0719aL.tsv",
size = 10, sample_name = "E10_Eye_2", image_name = "main")VoltRon Object
E10_Eye_2:
Layers: Section1
Assays: DBIT-Seq-RNA(Main) DBIT-Seq-Prot ST data
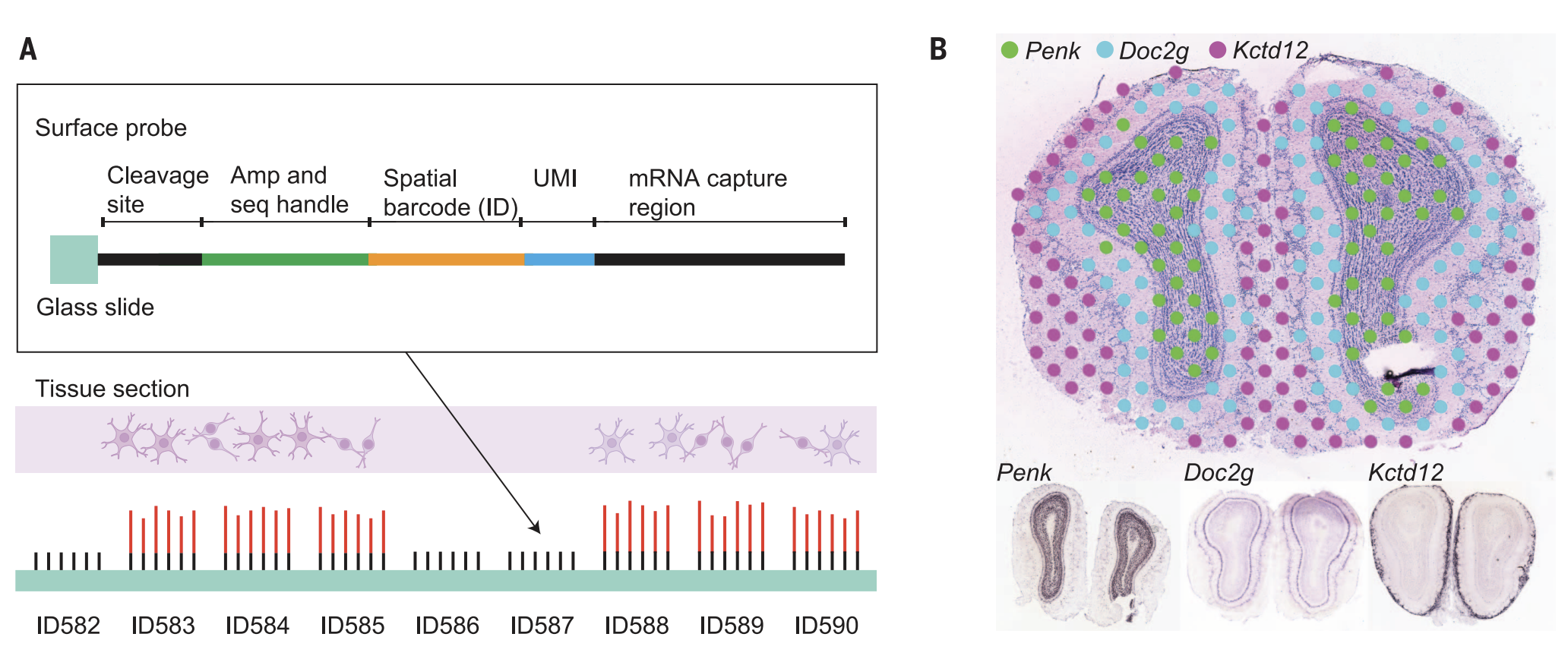
Image Credit: Ståhl, et. al (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science, 353(6294), 78-82.
We demonstrate importing the original Spatial Transcriptomics (ST) datasets by formulating custom spot-level spatial transcriptomics datasets. We will use the formVoltRon function directly. We use the example provided by the https://doi.org/10.5281/zenodo.4751624. For more information you can also check the paper here.
We first import and manipulate count matrices and spot coordinates from the provided output files. Here we only demonstrate building a custom spot-level VoltRon object for the section “A1”, however the remaining sections can be imported using the same method.
# count matrix
raw.data <- read.table("A1.tsv", header = TRUE, sep = "\t")
spatialentities <- raw.data$X
raw.data <- raw.data[,-1]
raw.data <- t(as.matrix(raw.data))
# coords
coords <- read.table("A1_selection.tsv", header = TRUE, sep = "\t")
rownames(coords) <- paste(coords$x, coords$y, sep = "x")
coords <- coords[spatialentities,]
coords <- coords[,c("pixel_x", "pixel_y")]
colnames(coords) <- c("x", "y")The image can be imported using the magick package.
library(magick)
img <- magick::image_read("HE/A1.jpg")
img_info <- magick::image_info(img)Before we form the VoltRon object, we should define the parameters for spot-level datasets. Here, we provide
- the radius of a spot in the physical space (i.e. spot.radius)
- the radius of a spot for visualization (i.e. vis.spot.radius)
- The distance to the nearest spot, to be used for spatial neighborhood calculation (i.e. nearestpost.distance)
The scaling parameter (scale_param) is required to overlay localization and distances of spots to the coordinate system of the imported image. Here,
- the width of a ST slide is of 6200\(\mu\)m,
- the diameter of a spot is 100\(\mu\)m (hence a radius of 50\(\mu\)m) and
- the center-to-center distance between two spots is 200\(\mu\)m.
- Each spot has at most 8 neighboring spots including vertically, horizontally and diagonally adjacent spots.
scale_param <- img_info$width/6200
params <- list(
spot.radius = 50*(scale_param),
vis.spot.radius = 100*(scale_param),
nearestpost.distance = (200*sqrt(2) + 50)*scale_param
)Now we can combine all components into a VoltRon object. We should also flip the coordinates of spots vertically after creating the object.
# make voltron object
stdata <- formVoltRon(data = raw.data, image = img, coords = coords, assay.type = "spot",
params = params, sample_name = "A1")
stdata <- flipCoordinates(stdata)We can now visualize spots and the adjacency between these spots simultaneously.
stdata <- getSpatialNeighbors(stdata, method = "radius")
vrSpatialPlot(stdata, graph.name = "radius", crop = T)
Custom VoltRon objects
VoltRon incorporates the formVoltRon function to assemble each component of a spatial omics assay into a VoltRon object. Here:
- the feature matrix: the pxn feature to point matrix for raw counts and omics profiles
- metadata: the metadata table
- image: An image or a list of images with names associated to channel
- coordinates: xy-Coordinates of spatial points
- segments: the list of xy-Coordinates of each spatial point
can individually be prepared before executing formVoltRon.
We will use a single image based proteomics assay to demonstrate building custom VoltRon objects. Specifically, we use cells characterized by multi-epitope ligand cartography (MELC) with a panel of 44 parameters. We use the already segmented cells on which expression of 43 protein features (excluding DAPI) were mapped to these cells.
VoltRon also provides support for imaging based proteomics assays. In this next use case, we analyze cells characterized by multi-epitope ligand cartography (MELC) with a panel of 44 parameters. We use the already segmented cells on which expression of 43 protein features (excluding DAPI) were mapped to these cells. You can download the files below here.
library(magick)
# feature x cell matrix
intensity_data <- read.table("intensities.tsv", sep = "\t")
intensity_data <- as.matrix(intensity_data)
# metadata
metadata <- read.table("metadata.tsv", sep = "\t")
# coordinates
coordinates <- read.table("coordinates.tsv", sep = "\t")
coordinates <- as.matrix(coordinates)
# image
library(magick)
image <- image_read("DAPI.tif")
# create VoltRon object
vr_object<- formVoltRon(data = intensity_data,
metadata = metadata,
image = image,
coords = coordinates,
main.assay = "MELC",
assay.type = "cell",
sample_name = "control_case_3",
image_name = "DAPI")
vr_objectVoltRon Object
control_case_3:
Layers: Section1
Assays: MELC(Main) VoltRon can store multiple images (or channels) associated with a single coordinate system.
library(magick)
image <- list(DAPI = image_read("DAPI.tif"),
CD45 = image_read("CD45.tif"))
vr_object<- formVoltRon(data = intensity_data,
metadata = metadata,
image = image,
coords = coordinates,
main.assay = "MELC",
assay.type = "cell",
sample_name = "control_case_3",
image_name = "MELC")These channels then can be interrogated and used as background images for spatial plots and spatial feature plots as well.
vrImageChannelNames(vr_object) Assay Layer Sample Spatial Channels
Assay1 MELC Section1 control_case_3 MELC DAPI,CD45You can extract each of these channels individually.
vrImages(vr_object, name = "MELC", channel = "DAPI")
vrImages(vr_object, name = "MELC", channel = "CD45")

|
|
Multiplex IF (QuPath)
VoltRon supports the analysis of clustering cells captured from multiplex immunofluorescence (IF) experiment that typically captured and stored in ome.tiff and/or qptiff files of high-resolution images. To process and segment single cells as well as quantifying intensity of protein markers, we can use QuPath software. Extensions of QuPath such as InstanSeg and StarDist could be used to process images, which we then import into VoltRon to build multiple IF assays.
QuPath allows you do select an ROI, and then the users can go to Extensions -> InstanSeg -> Run InstanSeg. The pane of InstanSeg let you choose the model for segmentation, device (mps and cuda for gpu support) and the channels to learn the segmentation and calculating features. Once complete, (i) the feature x segments (cell) matrix can be exported from Measurements -> Show Detection Measurements -> save* as a text file, (ii)** the segments are exported from Export objects as GeoJSON excluding measurements and exporting as FeatureCollection.
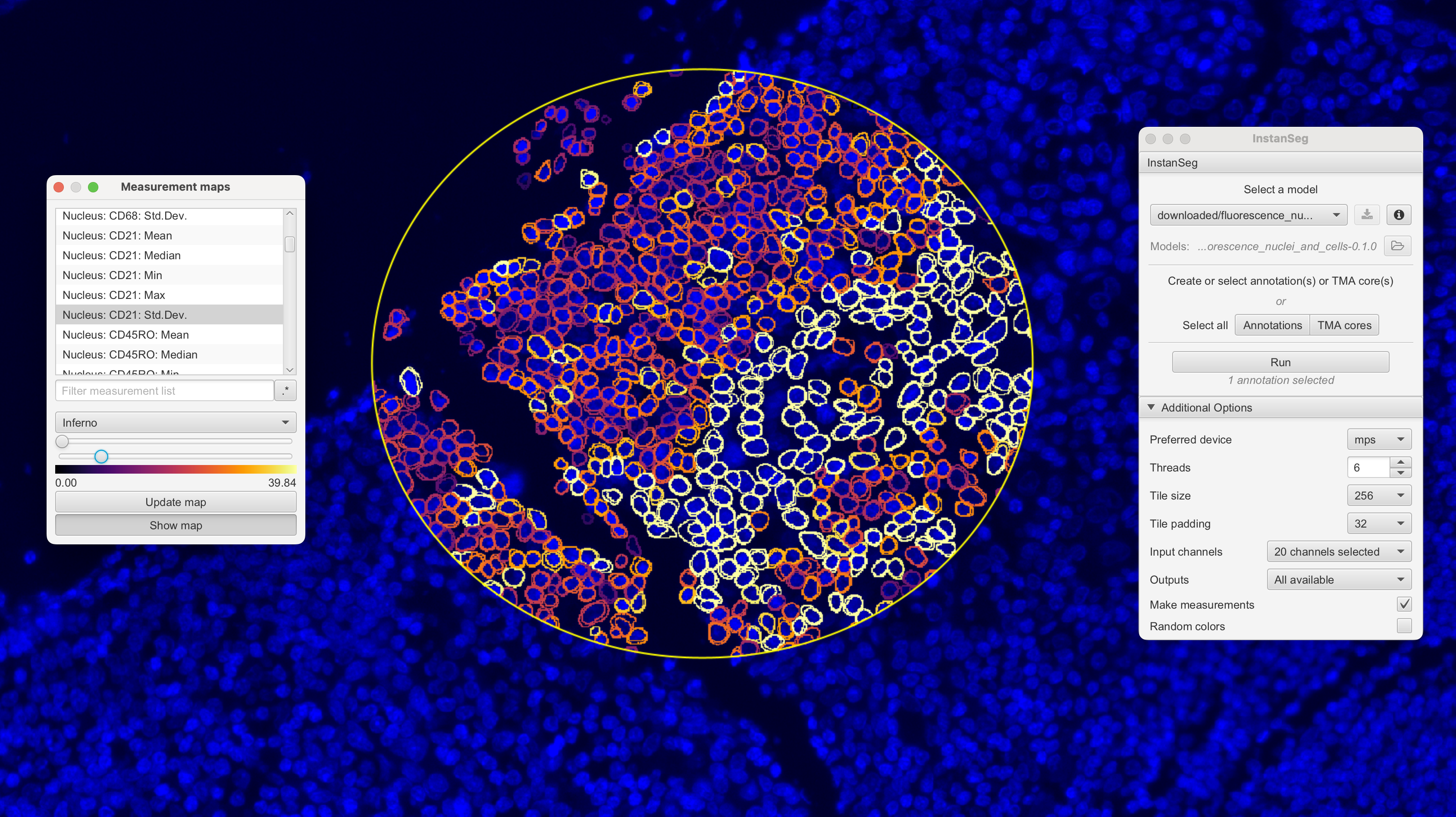
With these files, we can use the importQuPathIF function to import the data as a VoltRon object. You can select the resolution of the channels using series and resolution parameters which will pick the desired level from the ome.tiff file.
adj_vr <- importQuPathIF(measurements = "measurements.txt",
segments = "measurements.geojson",
image = "if_exp.ome.tif",
channels = c("DAPI", "CD20", "CD21"),
series = 1,
resolution = 1,
sample_name = "IFdata")You can use the following function to get the names of all channels in the ome.tiff file
getOmeTiffChannels(if_exp.ome.tif)Image-only VoltRon objects
With VoltRon we can also build objects where pixels (or tiles) are defined as spatial points. These information are derived from images only which then can be used for multiple downstream analysis purposes.
For this we incorporate importImageData function and only supply an image object. We will use the H&E image derived from a tissue section that was first analyzed by The 10x Genomics Xenium In Situ platform.
More information on the Xenium and the study can also be found on the bioRxiv preprint. You can download the H&E image from the 10x Genomics website as well (specifically, import the Post-Xenium H&E image (TIFF) data).
Xen_R1_image <- importImageData("Xenium_FFPE_Human_Breast_Cancer_Rep1_he_image.tif",
sample_name = "XeniumR1image",
image_name = "H&E")
Xen_R1_imageVoltRon Object
XeniumR1image:
Layers: Section1
Assays: ImageData(Main) vrImages(Xen_R1_image)
OME-TIFF
You can also import images from OME-TIFF files directly. Since these
files often come in shape of image
pyramids, we need to use RBioFormats the package.
The read.metadata function will allow you to browse the
existing resolutions and series in the image.
RBioFormats requires rJava and JDK installed, see dependencies section for more information.
We will use the post-Xenium OME-TIFF files from the Whole Mouse Pup slide from the 10X Genomics website. Please download the OME-TIFF file from output files.
# dependencies
if(!requireNamespace("RBioFormats"))
BiocManager::install("RBioFormats")
library(RBioFormats)
# import image
ome.tiff <- "Xenium_Prime_Mouse_Pup_FFPE_he_image.ome.tif"
read.metadata(ome.tiff)ImageMetadata list of length 10
series res sizeX sizeY sizeC sizeZ sizeT total
1 1 90959 62008 3 1 1 3
1 2 45479 31004 3 1 1 3
1 3 22739 15502 3 1 1 3
1 4 11369 7751 3 1 1 3
1 5 5684 3875 3 1 1 3
1 6 2842 1937 3 1 1 3
1 7 1421 968 3 1 1 3
1 8 710 484 3 1 1 3
1 9 355 242 3 1 1 3
1 10 177 121 3 1 1 3
globalMetadata:List of 1525
$ 20x_BF_01 Microscope Autoexposure enabled : chr "0"
$ 20x_BF_01 Value #359 : chr "(42753.440890479455, 8472.726448134907)"
$ 20x_BF_01 Value #358 : chr "-122.4"
$ 20x_BF_01 Units #049 : chr "10^-6m^1"
$ 20x_BF_01 Value #357 : chr "0"
[list output truncated]Now, we can specify the series and
resolution to select the image layer we want to import to
VoltRon.
vrimagedata <- importImageData(ome.tiff, series = 1, resolution = 5)
vrImages(vrimagedata)