Conversion
Conversion to Other Platforms
VoltRon is capable of end-to-end spatial data analysis for all levels of spatial resolutions, including those of single cell resolution. However, VoltRon provides a ecosystem friendly infrastructure where VoltRon objects could be transformed into data structures used by popular computational platforms such as Seurat, Squidpy and even Zarr for interactive spatial data visualization with Vitessce.
For both Seurat (R) and Squidpy (Python), we analyse readouts of the experiments conducted on example tissue sections analysed by the Xenium In Situ platform. For more information on processing and analyzing Xenium datasets, check the Cell/Spot Analysis tutorial.
Seurat
We will first see how we can transform VoltRon objects into Seurat object and use built-in functions such as FindAllMarkers to visualize marker genes of clusters found by VoltRon. You can find the clustered Xenium data using VoltRon here.
Xen_data <- readRDS("Xenium_data_clustered.rds")
SampleMetadata(Xen_data) Assay Layer Sample
Assay1 Xenium Section1 XeniumR1
Assay3 Xenium Section1 XeniumR2We use the as.Seurat function to convert spatial assays of VoltRon into Seurat objects. Here, a Seurat object defines spatial components of cellular and subcellular assays as FOV objects, and we use the type = “image” argument to convert spatial coordinates of cells and molecules into individual FOV objects for each Xenium assay/layer in the VoltRon object.
Please check the Analysis of Image-based Spatial Data in Seurat tutorial for more information on analyzing FOV-based spatial data sets with Seurat.
note: Use VoltRon::as.Seurat to avoid conflict with Seurat package’s as.Seurat function
library(Seurat)
Xen_data_seu <- VoltRon::as.Seurat(Xen_data, cell.assay = "Xenium", type = "image")
Xen_data_seu <- NormalizeData(Xen_data_seu)
Xen_data_seuAn object of class Seurat
313 features across 283298 samples within 1 assay
Active assay: Xenium (313 features, 0 variable features)
1 layers present: counts
2 dimensional reductions calculated: pca, umap
2 spatial fields of view present: fov_Assay1 fov_Assay3Marker Analysis
Now that we converted VoltRon into a Seurat object, we can pick the Clusters metadata column indicating the clustering of cells and test for marker genes of each individual cluster.
Idents(Xen_data_seu) <- "Clusters"
markers <- FindAllMarkers(Xen_data_seu)
head(markers[order(markers$avg_log2FC, decreasing = TRUE),]) p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
CPA3 0 7.343881 0.977 0.029 0 16 CPA3
CTSG 0 7.114698 0.878 0.011 0 16 CTSG
LILRA4.1 0 6.992717 0.939 0.015 0 19 LILRA4
ADIPOQ 0 6.860190 0.974 0.025 0 5 ADIPOQ
MS4A1 0 6.763083 0.919 0.027 0 17 MS4A1
BANK1 0 6.082192 0.889 0.037 0 17 BANK1Visualization
We can now pick top positive markers from each of these clusters prior to visualization.
library(dplyr)
topmarkers <- markers %>%
group_by(cluster) %>%
top_n(n = 5, wt = avg_log2FC)Here, VoltRon incorporates the unique markers learned by the FindAllMarkers function from Seurat and uses them to visualize the expression of these markers on heatmaps, and now we can also use these markers for annotating the clusters.
library(ComplexHeatmap)
marker_features <- unique(topmarkers$gene)
vrHeatmapPlot(Xen_data, features = marker_features, group.by = "Clusters",
show_row_names = TRUE, font.size = 10)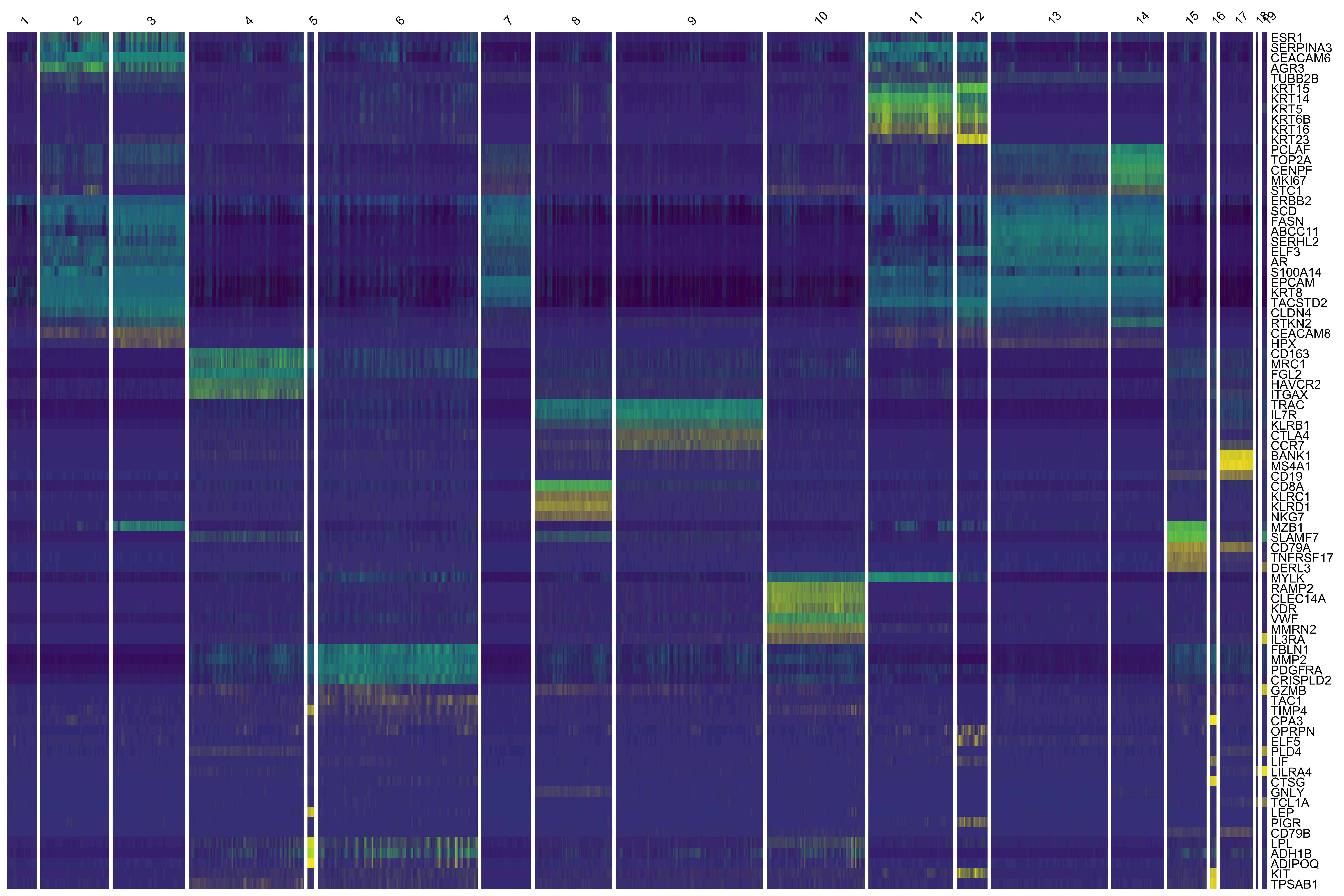
Convert with Molecule Data
If defined, the as.Seurat function may also convert the molecule assay of the VoltRon object into a Seurat FOV object and allow visualizing molecules. You can find the Xenium VoltRon object with the molecule assay here.
Xen_R1 <- readRDS("Xen_R1.rds")
SampleMetadata(Xen_R1) Assay Layer Sample
Assay1 Xenium Section1 XeniumR1
Assay2 Xenium_mol Section1 XeniumR1We define both the cell level assay and the molecule level assay.
Xen_R1_seu <- as.Seurat(Xen_R1, cell.assay = "Xenium", molecule.assay = "Xenium_mol", type = "image")Now we can visualize molecules alongside with cells.
ImageDimPlot(Xen_R1_seu, fov = "fovAssay1", molecules = "PGR", group.by = "orig.ident", cols = "lightgrey", mols.size = 1)
SpatialExperiment
VoltRon can also convert objects in SpatialExperiment objects. We are going to use the Xenium data clustered using VoltRon here.
Xen_data <- readRDS("Xenium_data_clustered.rds")
SampleMetadata(Xen_data)We use the as.SpatialExperiment function to convert spatial assays of VoltRon into SpatialExperiment objects. Please check the Introduction to the SpatialExperiment class tutorial for more information.
library(SpatialExperiment)
spe <- as.SpatialExperiment(Xen_data, assay = "Xenium")Here we can parse the image and visualize.
img <- imgRaster(spe,
sample_id = "Assay1",
image_id = "main")
plot(img)Squidpy (AnnData, h5ad)
A true ecosystem friendly computational platform should support data types across multiple computing environments. By allowing users to convert VoltRon objects into annotated data matrix formats such as AnnData, we can use built-in spatial data analysis methods available on squidpy.
You can find the clustered and the annotated Xenium data using VoltRon here.
AnnData objects wrapped on h5ad files are commonly used by the scverse ecosystem for single cell analysis which bring together numeruous tools maintained and distributed by a large community effort. Both squidpy and scanpy are currently available on scverse.
Xen_data <- readRDS("Xenium_data_clustered_annotated.rds")
as.AnnData(Xen_data, assay = "Xenium", file = "Xen_adata_annotated.h5ad")Configure Squidpy (scverse)
Here, we use the reticulate package to call scverse module in Python through a prebuilt anaconda environment. However, any python installation with the scverse module can be incorporated by reticulate.
library(reticulate)
use_condaenv("scverse", required = T)We import some other necessary modules such as pandas, scanpy and squidpy.
from pathlib import Path
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
import scanpy as sc
import squidpy as sq
sc.logging.print_header()Filter and Normalize
We read the annotated Xenium object that was saved as an h5ad file using the as.AnnData function in VoltRon, and process before analysis. For more information using scanpy and squidpy on Xenium datasets, check the Analyzing Xenium data tutorial at squidpy webpage.
adata = sc.read_h5ad("Xen_adata_annotated.h5ad")
adata.layers["counts"] = adata.X.copy()
sc.pp.normalize_total(adata, inplace=True)
sc.pp.log1p(adata)Visualize
We use the squidpy.pl.spatial_scatter functions available in squidpy to visualize the spatial localization of cell types of both Xenium replicates.
fig, ax = plt.subplots(1, 2, figsize=(10, 7))
sq.pl.spatial_scatter(adata, library_key = "library_id", library_id = "Assay1",
color=["CellType"], shape=None, size=1, img = False, ax=ax[0])
sq.pl.spatial_scatter(adata, library_key = "library_id", library_id = "Assay3",
color=["CellType"], shape=None, size=1, img = False, ax=ax[1])
plt.show(ax)
Neighborhood Enrichment
We can now use high level spatially-aware functions available in squidpy. We first establish spatial neighbors using the delaunay graphs. The spatial graph and distances will be stored under .obsp attribute/matrix.
sq.gr.spatial_neighbors(adata, coord_type="generic", delaunay=True)
adataAnnData object with n_obs × n_vars = 283298 × 313
obs: 'Count', 'Assay', 'Layer', 'Sample', 'Clusters', 'CellType', 'library_id'
uns: 'log1p', 'spatial_neighbors'
obsm: 'spatial'
layers: 'counts'
obsp: 'spatial_connectivities', 'spatial_distances'We can now conduct the permutation test for neighborhood enrichment across cell type pairs.
sq.gr.nhood_enrichment(adata, cluster_key="CellType")fig, ax = plt.subplots(1, 2, figsize=(13, 7))
sq.pl.nhood_enrichment(adata, cluster_key="CellType", figsize=(8, 8),
title="Neighborhood enrichment adata", ax=ax[0])
sq.pl.spatial_scatter(adata, color="CellType", library_key = "library_id",
library_id = "Assay1", shape=None, size=2, ax=ax[1])
plt.show(ax)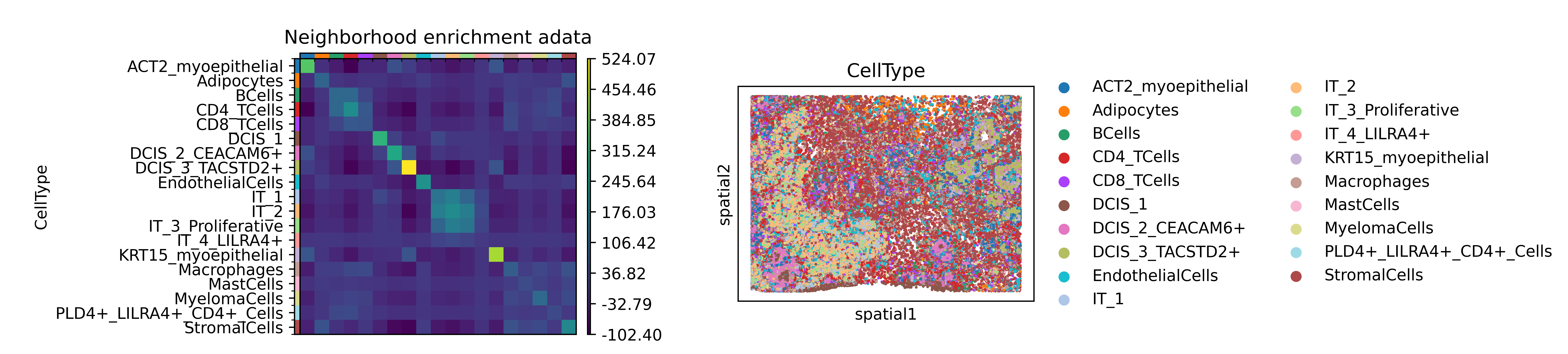
Vitessce (AnnData, zarr)
In this section, we will transform VoltRon objects of Xenium data into zarr arrays, and use them for interactive visualization in Vitessce. We should first download the vitessceR package which incorporates wrapper function to visualize zarr arrays interactively in R.
install.packages("devtools")
devtools::install_github("vitessce/vitessceR")You can find the clustered and annotated Xenium data using VoltRon here.
Xen_data <- readRDS("Xenium_data_clustered_annotated.rds")
SampleMetadata(Xen_data) Assay Layer Sample
Assay1 Xenium Section1 XeniumR1
Assay2 Xenium Section1 XeniumR2Interactive Visualization
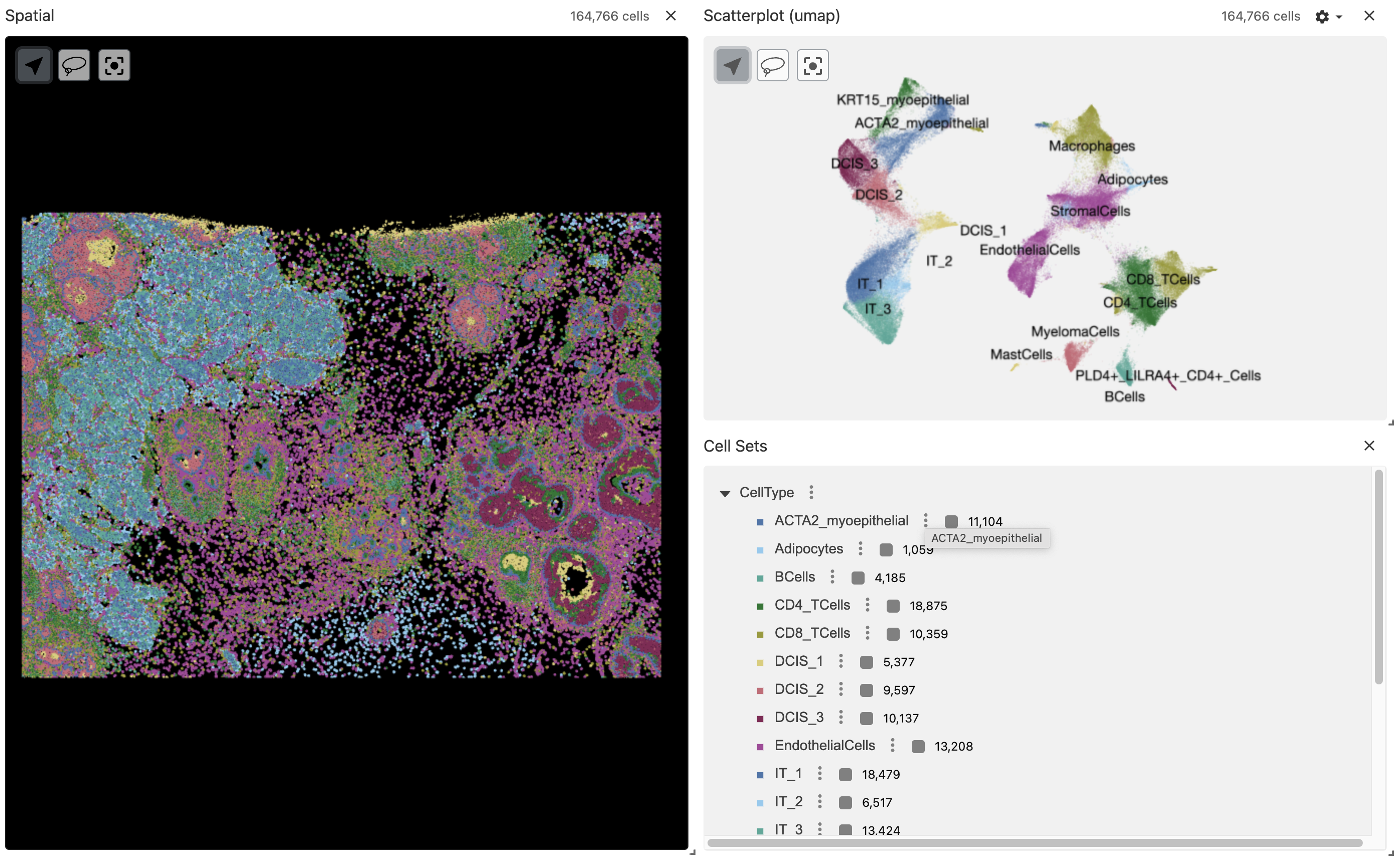
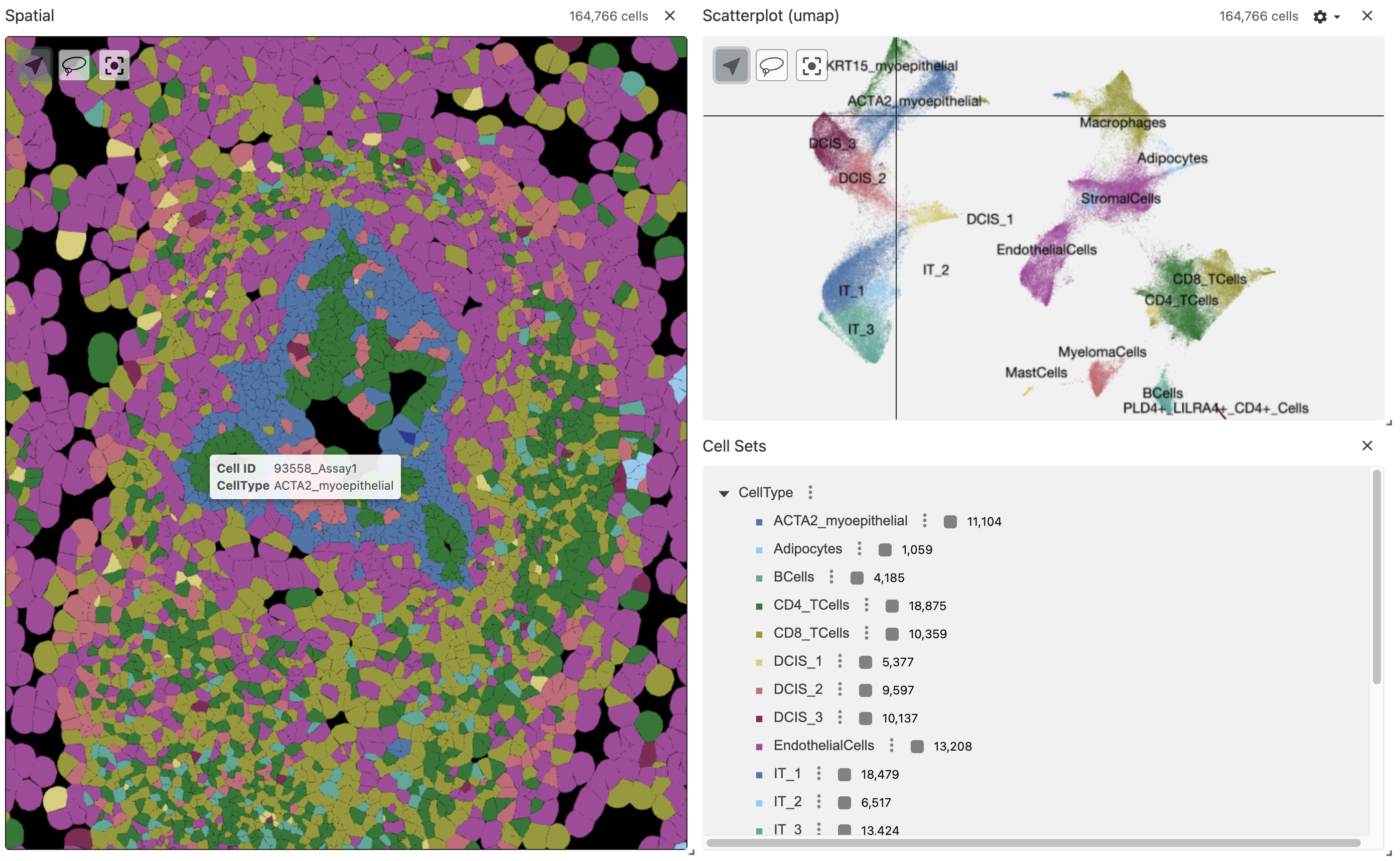
Now we can convert the VoltRon object into a zarr array using the as.AnnData function which will create the AnnData object as a zarr array in the specified location.
as.AnnData(Xen_data, assays = "Assay1",
file = "xendata_clustered_annotated.zarr", flip_coordinates = TRUE)We can use the zarr file directly in the vrSpatialPlot function to visualize the zarr array interactively in RStudio viewer. The reduction arguement allows the umap of the Xenium data to be visualized alongside with the spatial coordinates of the Xenium cells.
vrSpatialPlot("xendata_clustered_annotated.zarr", group.by = "CellType", reduction = "umap")