PiGx RNA-seq
Introduction
PiGx RNAseq is a preprocessing and analysis pipeline. It takes single-end and/or paired-end fastq files containing fragment reads, and does all the necessary preprocessing to get analysis-ready gene expression levels. It also performs quality control steps and outputs comprehensive quality statistics. Finally, it performs a differential expression analysis and outputs a stand-alone HTML report with tables and figures summarizing any differential gene expression between samples as specified in the experiment design.
Workflow
PiGx RNAseq follows academic best practices for preprocessing and analysis of RNAseq data. Figure 1 provides an overview of the different steps of the pipeline, as well as the outputs.
First, raw reads are trimmed using TrimGalore! to ensure a minimum read quality, and removal of adapter sequences. Next, reads are aligned to a reference genome using STAR, and the depth of coverage (normalized by DESeq2 size factors), is computed using GenomicAlignments, outputing bigwig files. Gene-level expression counts is obtained from GenomicAlignments::summarizeOverlaps, and transcript-level quantification is produced using Salmon. Statistical analysis for differential expression analysis is performed using DESeq2, GO term enrichment analysis is performed using gProfileR, and the results are used to compile a custom report.
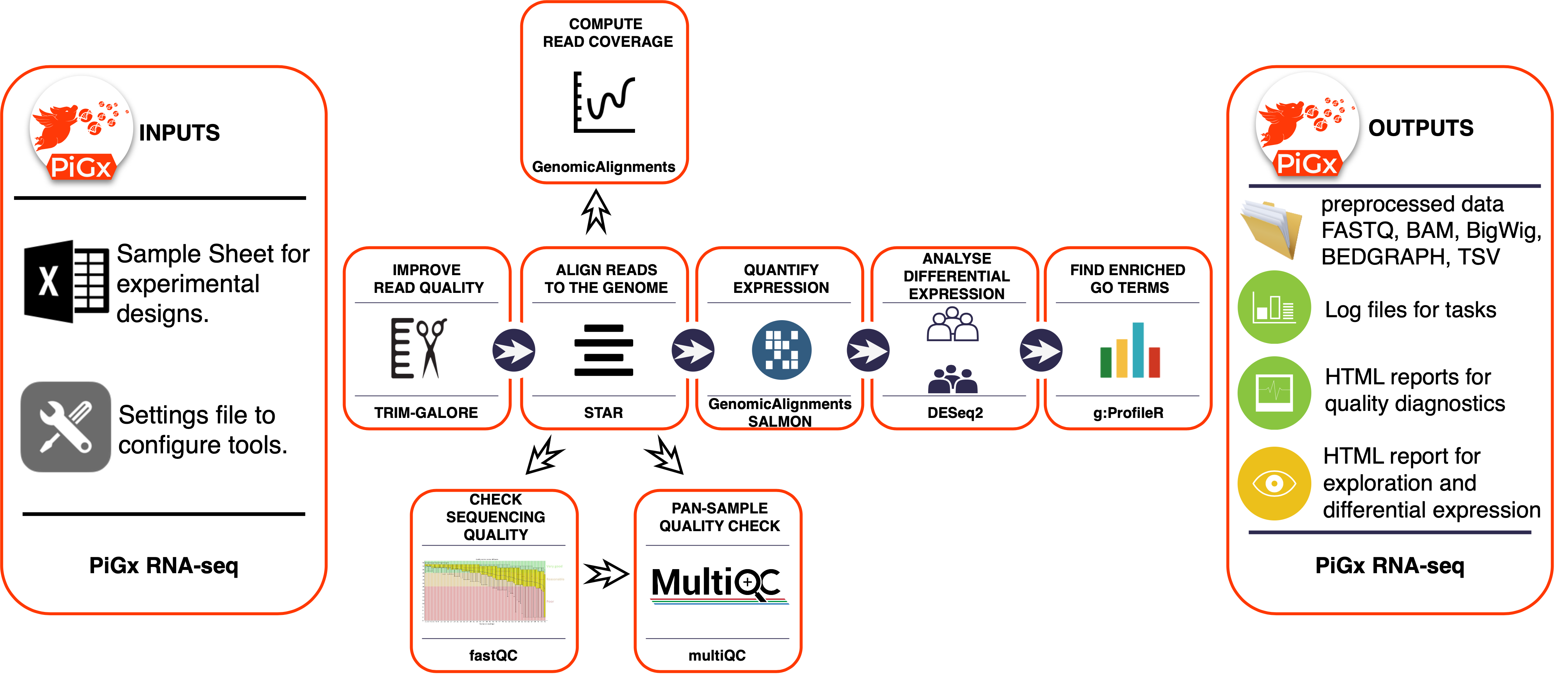 Figure 1: An overview of the PiGx RNAseq workflow
Figure 1: An overview of the PiGx RNAseq workflow
Output
- Quality Control reports
- Alignment results in BAM file format.
- bigwig files for genome-scale normalized coverage tracks
- Raw (via Salmon and STAR) and normalized read count tables (using DESeq2 median of ratios normalization procedure and TPM normalization).
- Differential expression analysis results
- HTML reports
- Tab-separated file for log-transformed normalized counts
- GO term analysis result tables
- DESeq2 differential expression results table sorted by adjusted p-values.
Installation
Pre-built binaries for PiGx are available through GNU Guix, the functional package manager for reproducible, user-controlled software management. Install the complete pipeline bundle with the following command:
guix install pigxYou can also install just the RNAseq pipeline with
guix install pigx-rnaseqIf you want to install PiGx RNAseq from source, please make sure that all required dependencies (see “requirements.txt”) are installed and then follow the common GNU build system steps after unpacking the latest release tarball:
./configure --prefix=/some/where
make installQuick start
Download the zipped test data folder from here:
Unzip the archive
tar -xzvf test_data.tgzChange to the test_data folder
cd test_dataRun the pipeline
pigx rnaseq -s ./settings.yaml ./sample_sheet.csv
Preparing the input
In order to run the pipeline, the user must supply
- a sample sheet
- a settings file
both files are described below.
In order to generate template settings and sample sheet files, type
pigx rnaseq --initin the shell, and a boilerplate sample_sheet.csv and settings.yaml will be written to your current directory.
Sample sheet
The sample sheet is a tabular file (csv format) describing the experiment. The table has the following columns:
| name | reads | reads2 | sample_type | (covariate1) | … | (covariateN) |
|---|---|---|---|---|---|---|
| ctrl | ctrl_R1.fastq.gz | ctrl_R2.fastq.gz | control | . | . | . |
| trtA | trtA_R1.fastq.gz | trtA_R2.fastq.gz | treatment A | . | . | . |
Table 1: example sample sheet
Column descriptions
- name is the name for the sample, which must be unique to each row of the table.
- reads1/2 are the fastq file names of paired end reads
- the location of these files is specified in
settings.yaml - for single-end data, leave the reads2 column in place, but have it empty
- the location of these files is specified in
- sample_type can be anything. For instance, a group of biologial replicates.
Additional columns may be included which may be used as covariates in the differential expression analysis (sex, age, batch).
Settings file
The settings file is a YAML file which specifies:
Locations:
- The locations of the reads (directory where
fastqfiles are) - The location of the outputs for the pipeline
- The location of the
fastafile with the reference genome (must be prepared by the user) - The locations of the transcriptome assembly (for alignment with salmon)
- The location of a
GTFfile with genome annotations
Organism (for GO-term analysis using gProfileR):
- Just append the species name to the initial of the genus name of the organism (e.g. hsapiens, mmusculus, celegans, dmelanogaster).
Differential Expression (DE) analyses to perform:
- Which samples to compare (by
sample_typein the sample sheet) - Which covariates to include in the DE analysis (from additional columns in the sample sheet)
In order to get started, enter pigx-rnaseq --init=settings my_settings.yaml. This will create a file called my_settings.yaml with the default structure. The file will look like this:
locations:
reads-dir: sample_data/reads/
output-dir: output/
genome-fasta: sample_data/sample.fasta
cdna-fasta: sample_data/sample.cdna.fasta
gtf-file: sample_data/sample.gtf
organism: hsapiens
DEanalyses:
#names of analyses can be anything but they have to be unique for each combination of case control group comparisons.
analysis1:
#if multiple sample names are provided, they must be separated by comma
case_sample_groups: "HBR"
control_sample_groups: "UHR"
covariates: ''
execution:
submit-to-cluster: no
jobs: 6
nice: 19Reference and annotations
The user must supply paths pointing to a reference genome for the organism, as well as annotated genes and a transcriptome reference. These might for example be downloaded from Ensembl. The reference genomoe is listed under DNA FASTA, the gene annotations under Gene sets GTF, and the transcriptome reference sequence is specified under cDNA FASTA. The user is free to choose any resource for these annotation files, however it is very important to note that the chromosome naming styles must match between different annotation files (e.g. chromosome 21 is named as chr21 in UCSC style, but 21 in NCBI/Ensemble style).
Organism (for GO-term analysis)
GO term analysis is performed based on the organism specified in the settings file. Some popular organisms include hsapiens, mmusculus, celegans, dmelanogaster. The full list of organism IDs is available on the gProfiler documentation, here.
DEanalysis
The section named DEanalyses in the settings file allows the user to specify a number of differential expression analyses to be performed. In the example above, analysis1 will be the name of the only analysis specified. In that analysis, samples with sample_type HBR will be compared with those with sample_type UHR, with no covariates included in the analysis.
Using multiple sample_types as cases or controls
The user may include more than one sample_type as the controls or cases for any DE analysis by specifiying a comma-spearated list. For example, the following block specified an analysis which compares samples belonging to mut1 and mut2 to the WT samples:
DEanalyses:
two_cases_analysis:
case_sample_groups: "mut1,mut2"
case_control_groups: "WT"The same may be done to specify several sample_types for the controls.
Covariates in DE analysis
Any number of additional columns may be added to the sample sheet and used as covariates in the DE analysis. The following sample sheet includes a column indicating the sex of the sample:
| name | reads | reads2 | sample_type | sex |
|---|---|---|---|---|
| treatment1 | treatment1.r1.fastq | treatment1.r2.fastq | treatment | m |
| treatment2 | treatment2.r1.fastq | treatment2.r2.fastq | treatment | f |
| control1 | control1.r1.fastq | control1.r2.fastq | control | m |
| control2 | control2.r1.fastq | control2.r2.fastq | control | f |
With the use of the following block in the settings file, the sex will be used as a covariate in the differential expression analysis:
DEanalyses:
analysis_with_covariate:
case_sample_groups: "treatment"
case_control_groups: "control"
covariates: "sex"Multiple covariates may be specified by providing a comma-separated list, such as
covariates: "sex,age,smoking_history"Warning: It is important to be aware of a common error that is thrown by DESeq2 when additional covariates are listed. In some cases, one or more covariates contain redundant information, or perfectly confounded by other covariates that are used to construct a design formula. In such cases, DESeq2 will throw this error: “the model matrix is not full rank, so the model cannot be fit as specified.”. This error signals the user to re-consider the list of covariates used and eliminate those that are redundant from this list. For more information on this topic, please refer to the vignette from DESeq2, here.
Running the pipeline
PiGx RNAseq is executed using the command pigx-rnaseq -s settings.yaml sample_sheet.csv. See pigx-rnaseq --help for information about additional command line arguments.
The execution section of the settings file provides some control over the execution of the pipeline.
Local / cluster execution
The workflow may be executed locally (on a single computer), or, if a Sun Grid Engine-compatible HPC environment is available, supports cluster execution. In order to enable cluster execution, specify submit-to-cluster: yes in the settings file.
Parallel execution
If the workflow is run on a cluster, or a single computer with sufficient resources, some of the tasks may be computed in parallel. To specify the allowed level or parallelism, set the jobs setting under execution in the settings file. For instance,
execution:
submit-to-cluster: yes
jobs: 40in the settings file, will submit up to 40 simultaneous compute jobs on the cluster.
Output description
PiGx RNAseq creates an output folder, as specified in the settings file, that contains all of the following outputs.
Quality control
General quality control metrics are computed using FastQC and MultiQC. The MultiQC report is particularly useful, collating quality control metrics from many steps of the pipeline in a single html report, which may be found under the multiqc folder in the PiGx output folder.
Gene expression
PiGx RNAseq produces three variants of gene expression count matrices:
| Kind of count matrix | output location |
|---|---|
| Post-alignment reads-per-gene counts from GenomicAlignments::summarizeOverlaps | in the feature_counts/raw_counts directory |
| Pseudo-alignment reads-per-gene counts from Salmon | in the feature_counts/raw_counts directory |
| Pseudo-alignment reads-per-transcript counts from Salmon | in the feature_counts/raw_counts directory |
The read counting step using GenomicAlignments::summarizeOverlaps can be configured by modifying the counting section of the settings.yaml file. By default, the reads that align to the exon features are grouped together via gene_id assuming no strand-speficity, assuming the input files are in bam format ordered by alignment coordinates. A counting mode of Union is chosen by default.
counting:
counting_mode: "Union" # other options are "IntersectionStrict" and "IntersectionNotEmpty"
count_nonunique: TRUE # boolean (see inter.feature argument of summarizeOverlaps)
strandedness: "unspecific" # other options are "forward" and "reverse" for strand-specific read-counting
feature: "exon"
group_feature_by: "gene_id"
yield_size: 2000000 # how many reads to process at a time (this impacts memory consumption)The arguments in the counting section can be modified. To find out more about the arguments for read counting, please refer to the GenomicAlignments::summarizeOverlaps function.
Normalized counts tables
In order to enable comparison of gene/transcript expression across all samples outside of the context of differential expression analysis, PiGx RNAseq produces normalized counts tables using two normalizatoin procedures:
- DESeq2 (median of ratios) normalization (Recommended option)
feature_counts/normalized/deseq_normalized_counts.tsvfor gene-level normalized counts.
- TPM normalization
feature_counts/normalized/TPM_counts_from_SALMON.transcripts.tsvfor transcript-level normalized counts.feature_counts/normalized/TPM_counts_from_SALMON.genes.tsvfor gene-level normalized counts.
Differential expression analysis results
PiGx RNAseq produces differential expression reports for each comparison specified in the settings file, and using each of the expression quantification strategies specified above. I.e. for each contrast specified in the settings file, three reports will be produced; one based on counts-per-gene from GenomicAlignments::summarizeOverlaps computed from STAR alignments, one based on counts-per-gene from Salmon, and another based on counts-per-transcript from Salmon. Along with the HTML report will be produced two additional files per comparison: 1) a tab-separated file containing the DESeq2 differential expression results table, and 2) a tab-separated file containing the normalized expression values. Notice that these normalized values only apply in the context of the compared samples. In order to compare normalized values across all samples, please use the DESeq2 normalized count tables at feature_counts/normalized/deseq_normalized_counts.tsv.
The reports and complementary tab-separated files are all saved in the reports output directory.
Depth of coverage
PiGx RNAseq computes coverage depth from the STAR-alignment of the reads, using GenomicAlignments::export.bw. The resulting bigwig files are output in the bigwig_files output folder.
Troubleshooting
Execution on a cluster
Currently, PiGx only supports Sun Grid Engine for cluster execution. If you’re uncertain about your cluster, try typing qsub in the shell (Sun Grid Engine uses qsub to submit jobs).
Disappearing jobs on the cluster
PiGx RNAseq comes with sensible defaults for resource requests when running on a cluster, but based on the genome version and other parameters, these might not be sufficient and your cluster might terminate your jobs. The cluster resource requests may be overridden in the settings file. See etc/settings.yaml for a full list.
Questions
If you have further questions please e-mail: pigx@googlegroups.com or use the web form to ask questions https://groups.google.com/forum/#!forum/pigx/