anglemania Tutorial
Aaron Kollotzek
Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, GermanyBerlin Institute for Medical Systems Biology, Berlin, Germanyaaron.kollotzek@mdc-berlin.de
Vedran Franke
Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, GermanyBerlin Institute for Medical Systems Biology, Berlin, GermanyArtem Baranovskii
Helmholtz MunichAltuna Akalin
Max Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, GermanyBerlin Institute for Medical Systems Biology, Berlin, Germany2025-10-23
Source:vignettes/anglemania_tutorial.Rmd
anglemania_tutorial.RmdIntroduction
anglemania is a feature selection package that extracts genes from
multi-batch scRNA-seq experiments for downstream dataset integration.
The goal is to select genes that carry high biological information and
low technical noise between the batches. Those genes are extracted from
gene pairs that have an invariant and extremely narrow or wide angle
between their expression vectors. Conventionally, highly-variable genes
(HVGs) or sometimes all genes are used for integration tasks (https://www.nature.com/articles/s41592-021-01336-8).
While HVGs are a great and easy way to reduce the noise and
dimensionality of the data, we hypothesize that there are better ways to
select genes specifically for integration tasks. HVGs are sensitive to
batch effects because the variance is a function of both the technical
and biological variance. anglemania improves conventional usage of HVGs
for integration tasks, especially when the transcriptional difference
between cell types or cell states is subtle (showcased here using
simulated data generated using splatter
with de.facLoc and de.facScale set to 0.1,
which results in mild differences between “Groups”). The package can be
used on top of SingleCellExperiment
or Seurat
objects.
Under the hood, anglemania works with file-backed big matrices (FBMs) from the bigstatsr package for fast and memory efficient computation.
Simulation
We simulate a scRNA-seq dataset using Splatter with 4 batches and 3 cell types with subtle differences between cell types and rather big batch effects.
batch.facLoc <- 0.3
de.facLoc <- 0.1
nBatches <- 4
nGroups <- 3
nGenes <- 5000
groupCells <- 300
sce_raw <- splatSimulate(
batchCells = rep(groupCells * nGroups, nBatches),
batch.facLoc = batch.facLoc,
group.prob = rep(1 / nGroups, nGroups),
nGenes = nGenes,
batch.facScale = 0.1,
method = "groups",
verbose = FALSE,
out.prob = 0.001,
de.prob = 0.1, # mild
de.facLoc = de.facLoc,
de.facScale = 0.1,
bcv.common = 0.1,
seed = 42
)
sce <- sce_raw
assays(sce)## List of length 6
## names(6): BatchCellMeans BaseCellMeans BCV CellMeans TrueCounts countsUnintegrated data
Here we perform a standard workflow on the unintegrated data. When we perform clustering on the unintegrated data and visualize it in a UMAP, we can see that the clusters are driven by batch effects rather than cell types.
sce_unintegrated <- sce
# Normalization.
sce_unintegrated <- logNormCounts(sce_unintegrated)
# Feature selection.
dec <- modelGeneVar(sce_unintegrated)
hvg <- getTopHVGs(dec, prop = 0.1)
# PCA.
set.seed(1234)
sce_unintegrated <- scater::runPCA(
sce_unintegrated,
ncomponents = 50,
subset_row = hvg
)
# Clustering.
colLabels(sce_unintegrated) <- clusterCells(sce_unintegrated,
use.dimred = "PCA",
BLUSPARAM = NNGraphParam(cluster.fun = "louvain")
)
# Visualization.
sce_unintegrated <- scater::runUMAP(sce_unintegrated, dimred = "PCA")
patchwork::wrap_plots(
plotUMAP(sce_unintegrated, colour_by = "Batch") +
ggtitle("Unintegrated data, colored by Batch"),
plotUMAP(sce_unintegrated, colour_by = "Group") +
ggtitle("Unintegrated data, colored by Group")
)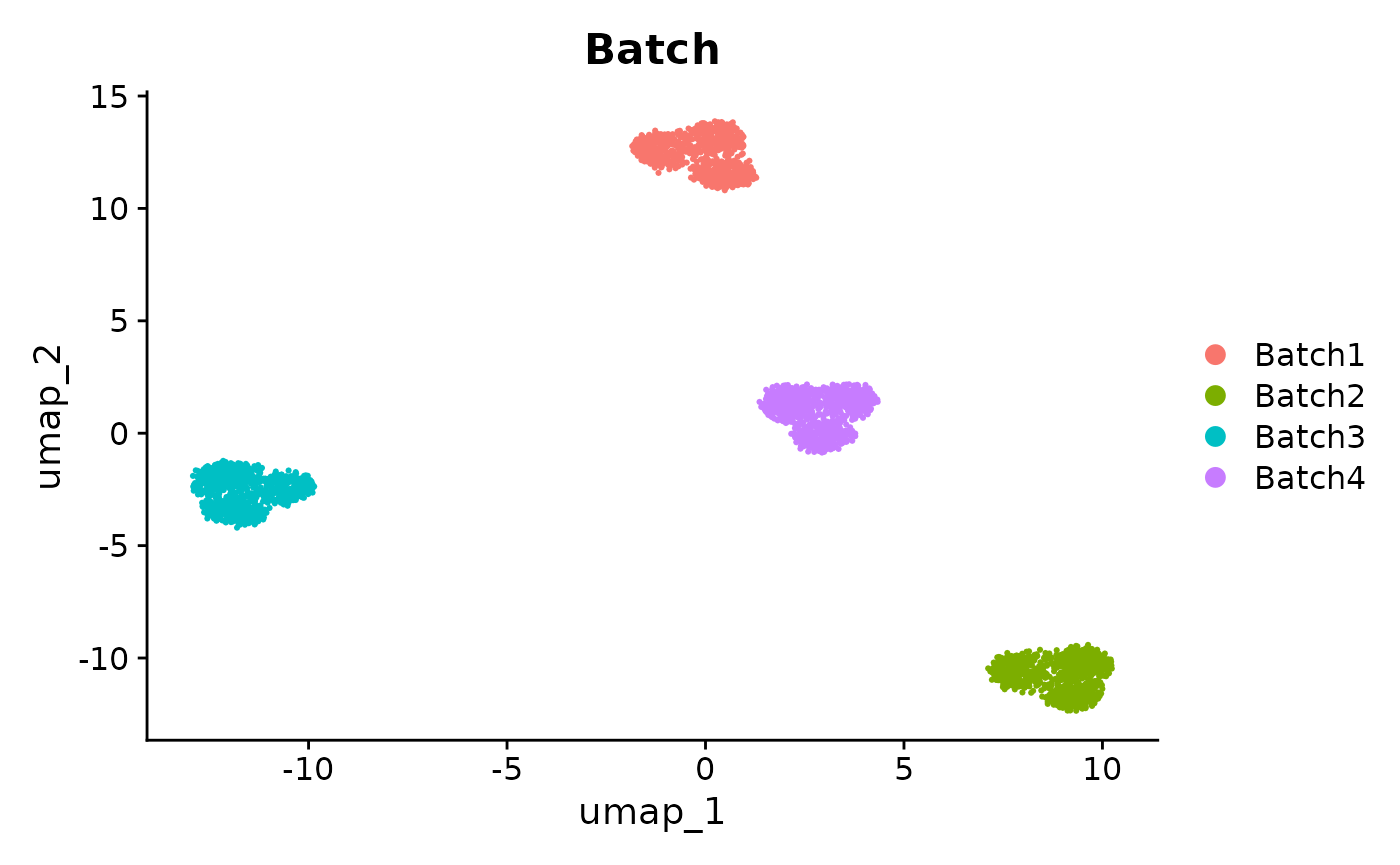
UMAPs of unintegrated data, colored by Batch and Group. The clusters are driven by batch effects.
anglemania
anglemania works on a SingleCellExperiment
object. The function has a few important arguments: -
batch_key: the column in the metadata of the
SingleCellExperiment object that indicates which batch the cells belong
to. This is required to distinguish between batches, because we compute
the angle between gene pairs for each batch. - method:
either cosine, spearman or diem - this is the method by which the
relationship of the gene pairs is measured. Default is cosine, which is
the cosine similarity between the expression vectors of the gene pairs.
- max_n_genes: you can specify a maximum number of
extracted genes. They are sorted by a weighted sum of the ranked mean
and standard deviation of the zscores of the gene pair correlations.
## DataFrame with 6 rows and 4 columns
## Cell Batch Group ExpLibSize
## <character> <character> <factor> <numeric>
## Cell1 Cell1 Batch1 Group1 46898.1
## Cell2 Cell2 Batch1 Group1 54688.2
## Cell3 Cell3 Batch1 Group2 52027.9
## Cell4 Cell4 Batch1 Group1 52319.5
## Cell5 Cell5 Batch1 Group3 37774.7
## Cell6 Cell6 Batch1 Group3 58112.3
batch_key <- "Batch"
sce <- anglemania(
sce,
batch_key = batch_key,
max_n_genes = 2000
)extract the anglemania genes from the SCE object
anglemania_genes <- get_anglemania_genes(sce)
head(anglemania_genes)## [1] "Gene527" "Gene4620" "Gene2249" "Gene3115" "Gene2802" "Gene3566"
length(anglemania_genes)## [1] 2000select_genes
Once anglemania was run on the SCE, you can adjust the number of
genes you want to select using the select_genes() function.
It has an argument score_weights, which let’s you decide how much weight
to set to the mean and standard deviation of the zscores. The default
which is used in the anglemania function is
0.4 for the mean zscore and 0.6 for the
standard deviation of the zscore. Higher weights for the standard
deviation of the zscores lead to a greater emphasis on genes with highly
stable correlation patterns.
sce <- select_genes(
sce,
max_n_genes = 2000,
score_weights = c(0.4, 0.6)
)
# Inspect the anglemania genes
anglemania_genes <- get_anglemania_genes(sce)
head(anglemania_genes)## [1] "Gene527" "Gene4620" "Gene2249" "Gene3115" "Gene2802" "Gene3566"
length(anglemania_genes)## [1] 2000MNN integration
The anglemania genes can now be used for downstream integration algorithms such as MNN. We compare the integration results using the anglemania genes with the results using 500 and the standard 2000 HVGs.
HVGs
500 HVGs
hvg_500 <- sce %>%
scater::logNormCounts() %>%
modelGeneVar(block = colData(sce)[[batch_key]]) %>%
getTopHVGs(n = 500)
barcodes_by_batch <- split(rownames(colData(sce)), colData(sce)[[batch_key]])
sce_list <- lapply(barcodes_by_batch, function(x) sce[, x])
sce_mnn <- sce %>%
scater::logNormCounts()
sce_mnn <- batchelor::fastMNN(
sce_mnn,
subset.row = hvg_500,
k = 20,
batch = factor(colData(sce_mnn)[[batch_key]]),
d = 50
)
reducedDim(sce, "MNN_hvg_500") <- reducedDim(sce_mnn, "corrected")
sce <- scater::runUMAP(sce, dimred = "MNN_hvg_500", name = "umap_MNN_hvg_500")
# k is the number of nearest neighbours to consider when identifying MNNs2000 HVGs
hvg_2000 <- sce %>%
scater::logNormCounts() %>%
modelGeneVar(block = colData(sce)[[batch_key]]) %>%
getTopHVGs(n = 2000)
barcodes_by_batch <- split(rownames(colData(sce)), colData(sce)[[batch_key]])
sce_list <- lapply(barcodes_by_batch, function(x) sce[, x])
sce_mnn <- sce %>%
scater::logNormCounts()
sce_mnn <- batchelor::fastMNN(
sce_mnn,
subset.row = hvg_2000,
k = 20,
batch = factor(colData(sce_mnn)[[batch_key]]),
d = 50
)
reducedDim(sce, "MNN_hvg_2000") <- reducedDim(sce_mnn, "corrected")
sce <- scater::runUMAP(sce, dimred = "MNN_hvg_2000", name = "umap_MNN_hvg_2000")anglemania genes
sce_mnn <- sce %>%
scater::logNormCounts()
sce_mnn <- batchelor::fastMNN(
sce_mnn,
subset.row = anglemania_genes,
k = 20,
batch = factor(colData(sce_mnn)[[batch_key]]),
d = 50
)
reducedDim(sce, "MNN_anglemania_2000") <- reducedDim(sce_mnn, "corrected")
sce <- scater::runUMAP(
sce,
dimred = "MNN_anglemania_2000",
name = "umap_MNN_anglemania_2000"
)
sce_mnn <- sce %>%
scater::logNormCounts()
sce_mnn <- batchelor::fastMNN(
sce_mnn,
subset.row = anglemania_genes[1:500],
k = 20,
batch = factor(colData(sce_mnn)[[batch_key]]),
d = 50
)
reducedDim(sce, "MNN_anglemania_500") <- reducedDim(sce_mnn, "corrected")
sce <- scater::runUMAP(
sce,
dimred = "MNN_anglemania_500",
name = "umap_MNN_anglemania_500"
)Plot
UMAP embeddings
We can see from the UMAPs that anglemania genes yield the best integration in terms of clustering by cell type and mixing the batches. The goal of an integration and subsequent clustering should be to have low intra cluster variance and high inter cluster variance. This is at least true for most downstream scRNA-seq analyses where the goal is to e.g. differentiate between cell types or cell states and annotate these.
# Use wrap_plots
patchwork::wrap_plots(
plotReducedDim(sce, colour_by = "Batch", dimred = "umap_MNN_anglemania_500") +
ggtitle("MNN integration using 500 anglemania genes, colored by Batch"),
plotReducedDim(sce, colour_by = "Group", dimred = "umap_MNN_anglemania_500") +
ggtitle("MNN integration using anglemania genes, colored by Group"),
plotReducedDim(sce, colour_by = "Batch", dimred = "umap_MNN_hvg_500") +
ggtitle("MNN integration using top 500 HVGs, colored by Batch"),
plotReducedDim(sce, colour_by = "Group", dimred = "umap_MNN_hvg_500") +
ggtitle("MNN integration using top 500 HVGs, colored by Group"),
ncol = 2
)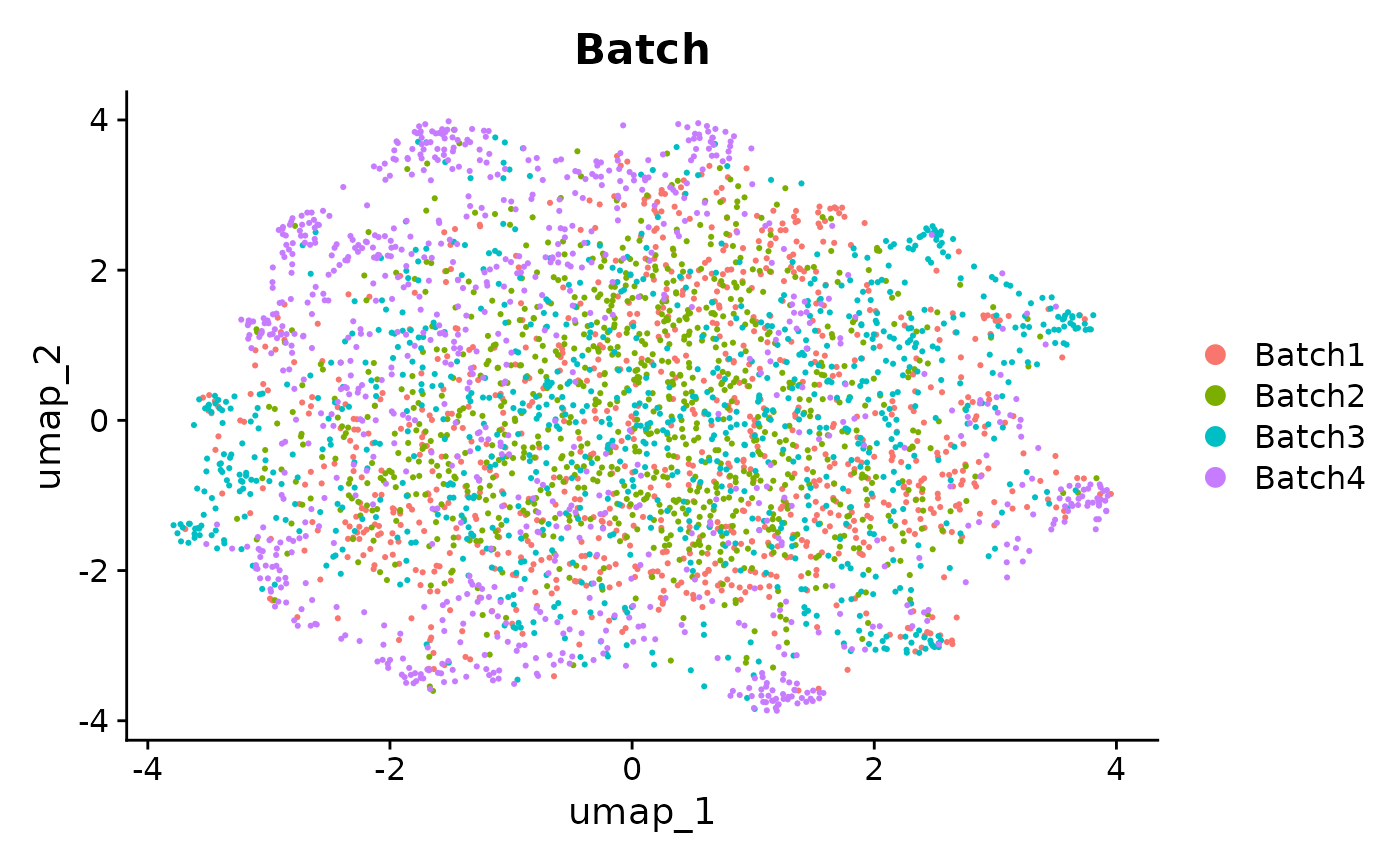
UMAPs of MNN integrated data. Comparison of UMAP embeddings of integrated data using anglemania genes, top 500 HVGs and top 2000 HVGs.
patchwork::wrap_plots(
plotReducedDim(sce, colour_by = "Batch", dimred = "umap_MNN_anglemania_2000") +
ggtitle("MNN integration using 2000 anglemania genes, colored by Batch"),
plotReducedDim(sce, colour_by = "Group", dimred = "umap_MNN_anglemania_2000") +
ggtitle("MNN integration using 2000 anglemania genes, colored by Group"),
plotReducedDim(sce, colour_by = "Batch", dimred = "umap_MNN_hvg_2000") +
ggtitle("MNN integration using top 2000 HVGs, colored by Batch"),
plotReducedDim(sce, colour_by = "Group", dimred = "umap_MNN_hvg_2000") +
ggtitle("MNN integration using top 2000 HVGs, colored by Group"),
ncol = 2
)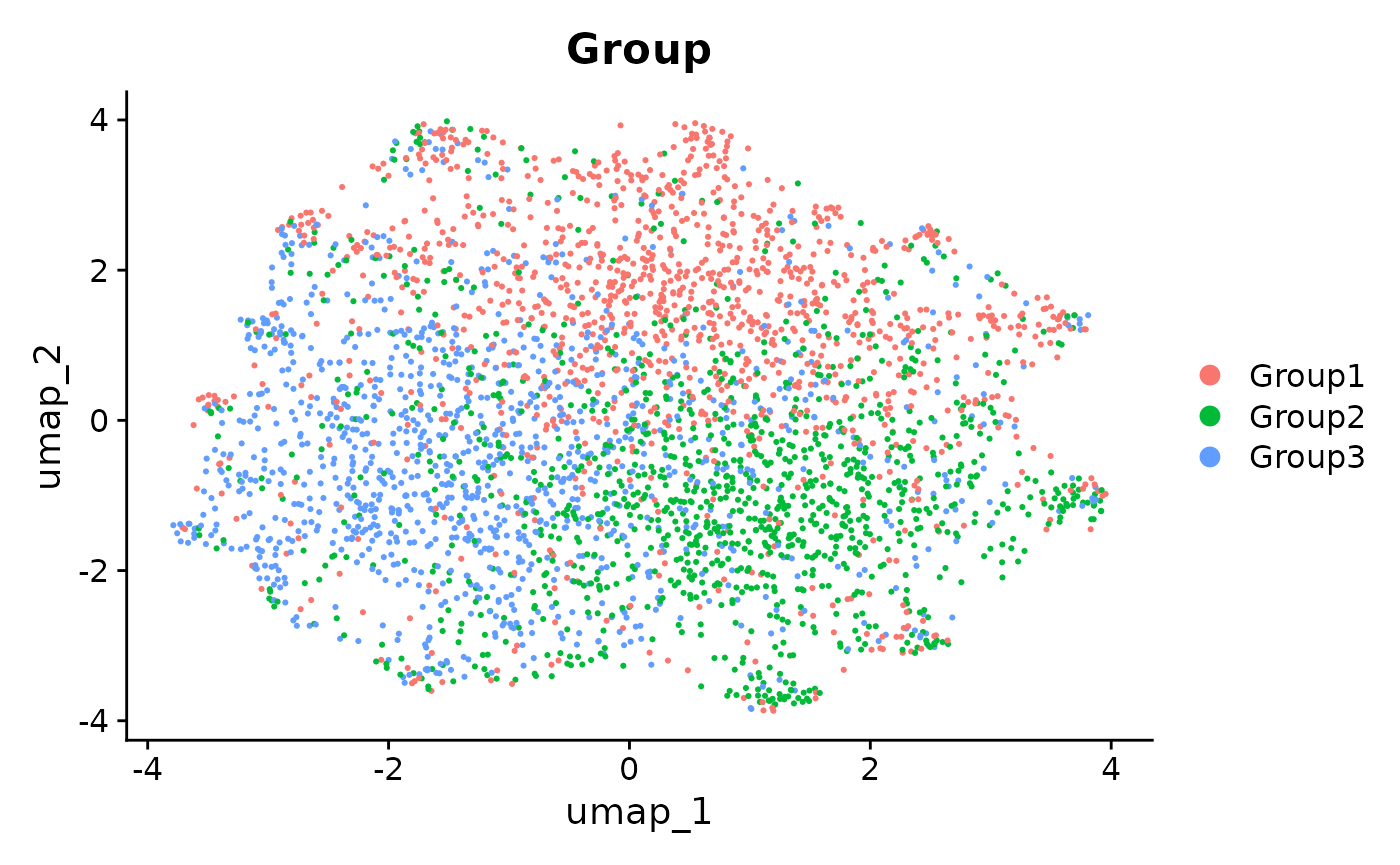
UMAPs of MNN integrated data. Comparison of UMAP embeddings of integrated data using anglemania genes, top 500 HVGs and top 2000 HVGs.
Overlap
upsetr_df <- fromList(
list(
anglemania = anglemania_genes,
hvg_500 = hvg_500,
hvg_2000 = hvg_2000
)
)
upset(upsetr_df, text.scale = 2)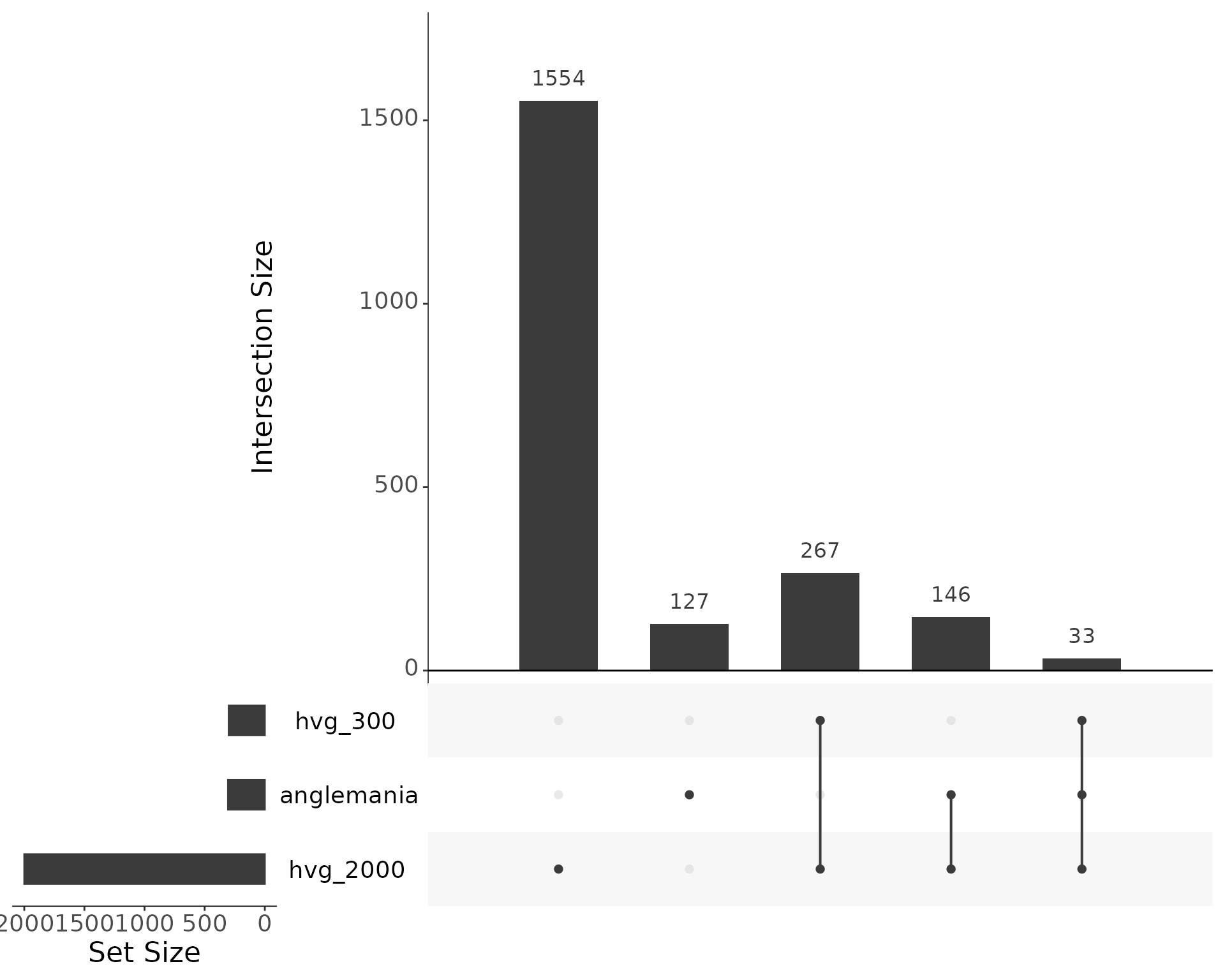
Overlap of selected genes. Additionally, we check the overlap of the anglemania genes with the HVGs. About 211 of the 2000 anglemania genes are also found in the top 500 HVGs, and 822 of the 2000 anglemania genes are also found in the top 2000 HVGs.
Seurat
Now you can just use the anglemania genes for other integration algorithms. When using Seurat, the easiest approach is to create an SCE from the counts and metadata of the SeuratObject, then run anglemania on it and save those genes as the VariableFeatures of the SeuratObject.
se <- CreateSeuratObject(
counts = counts(sce_raw),
meta.data = as.data.frame(colData(sce_raw))
)
se## An object of class Seurat
## 5000 features across 3600 samples within 1 assay
## Active assay: RNA (5000 features, 0 variable features)
## 1 layer present: counts
anglemania_genes <- se |>
as.SingleCellExperiment(assay = "RNA") |>
anglemania(
batch_key = "Batch",
max_n_genes = 2000
) |>
get_anglemania_genes()Integration
In Seurat v5 you split the layers of an assay by batch and then run
the normal Seurat workflow. To use anglemania genes for integration, you
need to assign them to the VariableFeatures slot of the SeuratObject.
After that, you integrate the layers using the anglemania genes as the
features argument.
# Split by batch
se[["RNA"]] <- split(se[["RNA"]], f = se$Batch)
# Standard preprocessing but use anglemania genes as "VariableFeatures"
se <- NormalizeData(se, verbose = FALSE)
VariableFeatures(se) <- anglemania_genes
se <- se |>
ScaleData(verbose = FALSE) |>
RunPCA(verbose = FALSE)
# Integrate
se <- IntegrateLayers(
object = se,
method = CCAIntegration,
orig.reduction = "pca",
new.reduction = "integrated.cca",
features = anglemania_genes,
verbose = FALSE
)
se <- RunUMAP(se, dims = 1:30, reduction = "integrated.cca", verbose = FALSE)
se## An object of class Seurat
## 5000 features across 3600 samples within 1 assay
## Active assay: RNA (5000 features, 2000 variable features)
## 9 layers present: counts.Batch1, counts.Batch2, counts.Batch3, counts.Batch4, data.Batch1, data.Batch2, data.Batch3, data.Batch4, scale.data
## 3 dimensional reductions calculated: pca, integrated.cca, umapPlot
patchwork::wrap_plots(
DimPlot(se, reduction = "umap", group.by = "Batch") +
ggtitle("Seurat integration using\nanglemania genes\ncolored by Batch"),
DimPlot(se, reduction = "umap", group.by = "Group") +
ggtitle("Seurat integration using\nanglemania genes\ncolored by Group"),
ncol = 2
)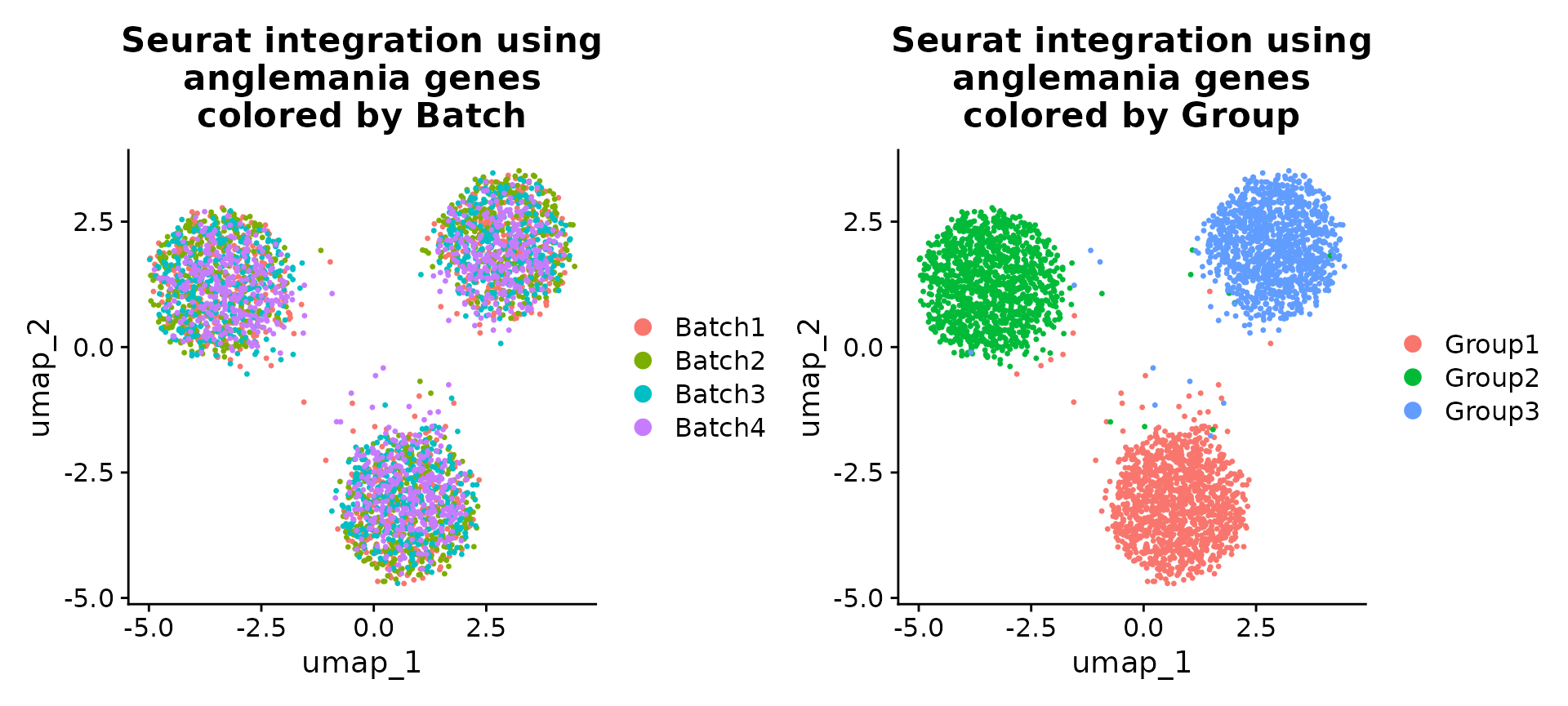
UMAPs of Seurat integrated data. Here we show that we can use the anglemania genes for integration of a SeuratObject.
Showcase underlying functions
Normal anglemania workflow
sce_raw <- sce_example()
sce <- sce_raw
batch_key <- "batch"
sce <- anglemania(sce, batch_key = batch_key, verbose = FALSE)anglemania is run on the SCE object and it basically
calls three functions:
-
factorise:- creates a permutation of the input matrix whose correlation matrix is used to create a null distribution for each batch.
- computes the cosine similarity (or spearman coefficient) between gene expression vector pairs matrix for both the original and permuted matrices
- computes the zscore of the relationship between the gene pairs taking the mean and standard deviation of the null distribution
- it does this for every batch in the dataset!
-
get_list_stats- computes the mean and standard deviation of the zscores across the
matrices from the different batches. This creates two important
matrices: the mean zscore matrix
mean_zscoreand the signal-to-noise ratio matrixsn_zscore. These are stored in the metadata of the SCE object.
- computes the mean and standard deviation of the zscores across the
matrices from the different batches. This creates two important
matrices: the mean zscore matrix
-
select_genes- filters the gene pairs by the
mean_zscoreandsn_zscorematrices (SN ratio, i.e. the mean divided by the standard deviation).
- filters the gene pairs by the
factorise
barcodes_by_batch <- split(rownames(colData(sce)), colData(sce)[[batch_key]])
counts_by_batch <- lapply(barcodes_by_batch, function(x) {
counts(sce[, x]) %>% sparse_to_fbm()
})
counts_by_batch[[1]][1:10, 1:6]## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 8 5 5 2 4 2
## [2,] 9 5 4 1 4 9
## [3,] 4 2 7 4 6 1
## [4,] 7 4 4 1 3 8
## [5,] 6 8 5 5 7 4
## [6,] 5 8 5 9 8 6
## [7,] 6 4 7 4 7 5
## [8,] 3 3 3 3 5 4
## [9,] 6 4 5 5 3 1
## [10,] 6 4 5 2 6 4
# we are working on FBMs (file-backed matrices
# implemented in the bigstatsr package)
class(counts_by_batch[[1]])## [1] "FBM"
## attr(,"package")
## [1] "bigstatsr"
# factorise produces the correlation matrices transformed to z-scores
factorised <- lapply(counts_by_batch, factorise)
factorised[[1]][1:10, 1:6]## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 0.00000000 -1.74668209 -0.79613304 1.1776966 0.8601212 0.50193905
## [2,] -1.73785984 0.00000000 0.59661885 2.5115045 0.5348975 1.43483620
## [3,] -0.85963025 0.53988067 0.00000000 0.2901283 -1.5767324 -0.06260101
## [4,] 1.13658651 2.45514150 0.33158301 0.0000000 -0.3715653 -0.57325460
## [5,] 0.92122274 0.57796947 -1.62420449 -0.3961608 0.0000000 -0.26750572
## [6,] 0.52925575 1.57028828 -0.02415793 -0.6582967 -0.2979901 0.00000000
## [7,] -0.41325531 -0.10548957 -1.16180200 0.0055108 0.3444741 1.41598180
## [8,] 0.10001406 -1.38192310 -1.29577006 -0.2524730 -0.7968541 -1.56575595
## [9,] 0.51996703 0.05963883 -0.37002326 -0.2927571 -0.9185203 -0.12159157
## [10,] 0.06970488 -0.33637803 1.57056534 0.3728527 0.5142178 1.01784989get_list_stats
The “list stats” are computed by get_list_stats and take
the z-score transformed correlation matrices from factorise
as input. The outputs are the mean zscore matrix
mean_zscore and the signal-to-noise ratio matrix
sn_zscore. These are stored in the metadata of the SCE
object.
matrix_list <- metadata(sce)$anglemania$matrix_list
weights <- setNames(
metadata(sce)$anglemania$params$dataset_weights$weight,
metadata(sce)$anglemania$params$dataset_weights$anglemania_batch
)
list_stats <- get_list_stats(
matrix_list = matrix_list,
weights = weights,
verbose = FALSE
)
names(list_stats)## [1] "mean_zscore" "sds_zscore" "sn_zscore"
class(list_stats)## [1] "list"
list_stats$mean_zscore[1:10, 1:6]## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 0.00000000 -1.4773877 -0.6690115 0.80957395 0.21191114 0.3065459
## [2,] -1.43297508 0.0000000 0.8094372 1.79591873 0.87491750 0.0903189
## [3,] -0.70975605 0.8090174 0.0000000 -0.90002758 -0.14215190 0.4217798
## [4,] 0.81025807 1.7916458 -0.8103351 0.00000000 -0.29386285 -0.2877692
## [5,] 0.26228504 0.8796571 -0.2190412 -0.31429554 0.00000000 -0.7944232
## [6,] 0.34210706 0.1727196 0.4235970 -0.32724467 -0.79625202 0.0000000
## [7,] 0.60813294 0.4576137 -1.4403958 -0.03807431 0.35694728 0.5394522
## [8,] -0.03868058 -1.0958225 -0.7680151 -0.62207485 -1.42724690 -0.8197054
## [9,] 1.26715940 0.3486664 -0.5030029 -0.76152650 -0.66668260 -0.4825756
## [10,] 0.49115807 1.1467226 0.7976727 -0.27013900 0.03028304 0.8112288
list_stats$sn_zscore[1:10, 1:6]## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] NA 3.87928890 3.7213414 1.5550667 0.23116551 1.1093569
## [2,] 3.3234406 NA 2.6894229 1.7746389 1.81948152 0.0475004
## [3,] 3.3486305 2.12554305 NA 0.5347330 0.07006687 0.6157208
## [4,] 1.7557127 1.90940919 0.5017815 NA 2.67420836 0.7127632
## [5,] 0.2814584 2.06177351 0.1102260 2.7147101 NA 1.0660911
## [6,] 1.2925884 0.08738832 0.6689559 0.6989744 1.12999842 NA
## [7,] 0.4210103 0.57464016 3.6559091 0.6177018 20.23533063 0.4351825
## [8,] 0.1972052 2.70836022 1.0290168 1.1901274 1.60093202 0.7769169
## [9,] 1.1991785 0.85301337 2.6746701 1.1487110 1.87190277 0.9452842
## [10,] 0.8240564 0.54672981 0.7297777 0.2970755 0.04424840 2.7762187
# Or we can access them directly from the SCE object
# after running anglemania
metadata(sce)$anglemania$list_stats$mean_zscore[1:10, 1:6]## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] 0.00000000 -1.4773877 -0.6690115 0.80957395 0.21191114 0.3065459
## [2,] -1.43297508 0.0000000 0.8094372 1.79591873 0.87491750 0.0903189
## [3,] -0.70975605 0.8090174 0.0000000 -0.90002758 -0.14215190 0.4217798
## [4,] 0.81025807 1.7916458 -0.8103351 0.00000000 -0.29386285 -0.2877692
## [5,] 0.26228504 0.8796571 -0.2190412 -0.31429554 0.00000000 -0.7944232
## [6,] 0.34210706 0.1727196 0.4235970 -0.32724467 -0.79625202 0.0000000
## [7,] 0.60813294 0.4576137 -1.4403958 -0.03807431 0.35694728 0.5394522
## [8,] -0.03868058 -1.0958225 -0.7680151 -0.62207485 -1.42724690 -0.8197054
## [9,] 1.26715940 0.3486664 -0.5030029 -0.76152650 -0.66668260 -0.4825756
## [10,] 0.49115807 1.1467226 0.7976727 -0.27013900 0.03028304 0.8112288
metadata(sce)$anglemania$list_stats$sn_zscore[1:10, 1:6]## [,1] [,2] [,3] [,4] [,5] [,6]
## [1,] NA 3.87928890 3.7213414 1.5550667 0.23116551 1.1093569
## [2,] 3.3234406 NA 2.6894229 1.7746389 1.81948152 0.0475004
## [3,] 3.3486305 2.12554305 NA 0.5347330 0.07006687 0.6157208
## [4,] 1.7557127 1.90940919 0.5017815 NA 2.67420836 0.7127632
## [5,] 0.2814584 2.06177351 0.1102260 2.7147101 NA 1.0660911
## [6,] 1.2925884 0.08738832 0.6689559 0.6989744 1.12999842 NA
## [7,] 0.4210103 0.57464016 3.6559091 0.6177018 20.23533063 0.4351825
## [8,] 0.1972052 2.70836022 1.0290168 1.1901274 1.60093202 0.7769169
## [9,] 1.1991785 0.85301337 2.6746701 1.1487110 1.87190277 0.9452842
## [10,] 0.8240564 0.54672981 0.7297777 0.2970755 0.04424840 2.7762187select_genes
- under the hood,
anglemaniacallsselect_genes - we can use
select_genesto change the number of selected genes without having to run anglemania again
previous_genes <- get_anglemania_genes(sce)
sce <- select_genes(
sce,
max_n_genes = 2000,
verbose = FALSE
)
# Inspect the anglemania genes
new_genes <- get_anglemania_genes(sce)
length(previous_genes)## [1] 300
length(new_genes)## [1] 300sessionInfo
## R version 4.5.1 (2025-06-13)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.3 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] future_1.67.0 UpSetR_1.4.0
## [3] batchelor_1.24.0 bluster_1.18.0
## [5] scran_1.36.0 scater_1.36.0
## [7] ggplot2_4.0.0 scuttle_1.18.0
## [9] splatter_1.32.0 SingleCellExperiment_1.30.1
## [11] SummarizedExperiment_1.38.1 Biobase_2.68.0
## [13] GenomicRanges_1.60.0 GenomeInfoDb_1.44.3
## [15] IRanges_2.42.0 S4Vectors_0.46.0
## [17] BiocGenerics_0.54.1 generics_0.1.4
## [19] MatrixGenerics_1.20.0 matrixStats_1.5.0
## [21] Seurat_5.3.0 SeuratObject_5.2.0
## [23] sp_2.2-0 dplyr_1.1.4
## [25] anglemania_0.99.7 BiocStyle_2.36.0
##
## loaded via a namespace (and not attached):
## [1] RcppAnnoy_0.0.22 splines_4.5.1
## [3] later_1.4.4 tibble_3.3.0
## [5] polyclip_1.10-7 fastDummies_1.7.5
## [7] lifecycle_1.0.4 edgeR_4.6.3
## [9] doParallel_1.0.17 globals_0.18.0
## [11] lattice_0.22-7 MASS_7.3-65
## [13] backports_1.5.0 magrittr_2.0.4
## [15] limma_3.64.3 plotly_4.11.0
## [17] sass_0.4.10 rmarkdown_2.30
## [19] jquerylib_0.1.4 yaml_2.3.10
## [21] bigparallelr_0.3.2 metapod_1.16.0
## [23] httpuv_1.6.16 otel_0.2.0
## [25] sctransform_0.4.2 spam_2.11-1
## [27] spatstat.sparse_3.1-0 reticulate_1.43.0
## [29] cowplot_1.2.0 pbapply_1.7-4
## [31] RColorBrewer_1.1-3 ResidualMatrix_1.18.0
## [33] abind_1.4-8 Rtsne_0.17
## [35] purrr_1.1.0 bigassertr_0.1.7
## [37] GenomeInfoDbData_1.2.14 ggrepel_0.9.6
## [39] irlba_2.3.5.1 listenv_0.9.1
## [41] spatstat.utils_3.2-0 goftest_1.2-3
## [43] RSpectra_0.16-2 dqrng_0.4.1
## [45] spatstat.random_3.4-2 fitdistrplus_1.2-4
## [47] parallelly_1.45.1 DelayedMatrixStats_1.30.0
## [49] pkgdown_2.1.3 codetools_0.2-20
## [51] DelayedArray_0.34.1 tidyselect_1.2.1
## [53] UCSC.utils_1.4.0 farver_2.1.2
## [55] viridis_0.6.5 ScaledMatrix_1.16.0
## [57] bigstatsr_1.6.2 spatstat.explore_3.5-3
## [59] flock_0.7 jsonlite_2.0.0
## [61] BiocNeighbors_2.2.0 progressr_0.17.0
## [63] ggridges_0.5.7 survival_3.8-3
## [65] iterators_1.0.14 systemfonts_1.3.1
## [67] foreach_1.5.2 tools_4.5.1
## [69] ragg_1.5.0 ica_1.0-3
## [71] Rcpp_1.1.0 glue_1.8.0
## [73] gridExtra_2.3 SparseArray_1.8.1
## [75] xfun_0.53 withr_3.0.2
## [77] BiocManager_1.30.26 fastmap_1.2.0
## [79] rsvd_1.0.5 digest_0.6.37
## [81] R6_2.6.1 mime_0.13
## [83] textshaping_1.0.4 scattermore_1.2
## [85] tensor_1.5.1 spatstat.data_3.1-9
## [87] tidyr_1.3.1 data.table_1.17.8
## [89] FNN_1.1.4.1 httr_1.4.7
## [91] htmlwidgets_1.6.4 S4Arrays_1.8.1
## [93] uwot_0.2.3 pkgconfig_2.0.3
## [95] gtable_0.3.6 lmtest_0.9-40
## [97] S7_0.2.0 XVector_0.48.0
## [99] htmltools_0.5.8.1 dotCall64_1.2
## [101] bookdown_0.45 scales_1.4.0
## [103] png_0.1-8 spatstat.univar_3.1-4
## [105] knitr_1.50 reshape2_1.4.4
## [107] checkmate_2.3.3 nlme_3.1-168
## [109] cachem_1.1.0 zoo_1.8-14
## [111] stringr_1.5.2 rmio_0.4.0
## [113] KernSmooth_2.23-26 vipor_0.4.7
## [115] parallel_4.5.1 miniUI_0.1.2
## [117] desc_1.4.3 pillar_1.11.1
## [119] grid_4.5.1 vctrs_0.6.5
## [121] RANN_2.6.2 promises_1.4.0
## [123] BiocSingular_1.24.0 ff_4.5.2
## [125] beachmat_2.24.0 xtable_1.8-4
## [127] cluster_2.1.8.1 beeswarm_0.4.0
## [129] evaluate_1.0.5 cli_3.6.5
## [131] locfit_1.5-9.12 compiler_4.5.1
## [133] rlang_1.1.6 crayon_1.5.3
## [135] future.apply_1.20.0 labeling_0.4.3
## [137] ps_1.9.1 ggbeeswarm_0.7.2
## [139] plyr_1.8.9 fs_1.6.6
## [141] stringi_1.8.7 viridisLite_0.4.2
## [143] deldir_2.0-4 BiocParallel_1.42.2
## [145] lazyeval_0.2.2 spatstat.geom_3.6-0
## [147] Matrix_1.7-3 RcppHNSW_0.6.0
## [149] patchwork_1.3.2 sparseMatrixStats_1.20.0
## [151] statmod_1.5.1 shiny_1.11.1
## [153] ROCR_1.0-11 igraph_2.2.0
## [155] bslib_0.9.0 bit_4.6.0